Полинейропатия — это патология периферической нервной системы, которая развивается в результате диффузного повреждения периферических нервов и их аксонов. Отсюда и название болезни. В ее основе — генерализованное поражение осевого цилиндра периферических нервов.

Что такое аксональная полинейропатия
Полиневропатия (второе название — полиневрит) — это клинический синдром, который возникает из-за ряда факторов, влияющих на периферическую нервную систему, и отличается размытыми патогенетическими изменениями. Заболевание занимает одно из лидирующих мест в перечне недугов периферической нервной системы, уступая первенство только вертеброгенной патологии, превосходящей по сложности клинической картины и последствиям, развивающимся из-за нее.
Аскональная полинейропатия считается междисциплинарной проблемой, с ней часто сталкиваются доктора различных специализаций. В первую очередь с данным заболеванием обращаются к неврологу. Частота возникающего синдрома неизвестна, так как отсутствуют статистические данные.
На данный момент известны всего три важных патоморфологических механизма, которые лежат в истоках формирования полинейропатии:
- валлеровская дегенерация;
- первичная демиелинизация;
- первичная аксонопатия.
В соответствии с иммунологической теорией полинейропатия является результатом перекрестного образования иммунных глобулинов, уничтожающих собственные клетки, в результате чего возникает некроз тканей и мышечное воспаление.
Исследователи выдвигают ряд гипотез возникновения и проблем течения аксональной полинейропатии:
- Сосудистая. Базируется на вовлечении в процесс сосудов, по которым кислород и питательные вещества поступают в периферические нервы. Изменяются характеристики крови по качественному и количественному составу, что может привести к ишемии нервных окончаний.
- Теория оксидативного стресса. Позиционирует формирование болезни со стороны нарушения обмена оксида азота, вследствие чего изменяются калий-натриевые механизмы, лежащие в основе формирования нервного возбуждения и проведения импульсов по нервам.
- Теория деактивации факторов роста нерва. Говорит о том, что болезнь возникает из-за недостатка аксонального транспорта с последующим развитием аксонопатии.
- Иммунологическая. Объясняет развитие заболевания в результате перекрестного образования антител к структурам периферической нервной системы, которое сопровождается аутоиммунным воспалением, а затем и некрозом нервов.
Даже при использовании ультрасовременных методов диагностики сложно найти достоверную причину патологии, выяснить ее получается только у 50-70% пострадавших.
Факторов возникновения полинейропатии нижних конечностей по аксональному типу очень много. Однако даже инновационные способы исследования не позволяют установить истинную этиологию заболевания.
Мнение эксперта
Автор: Алексей Владимирович Васильев
Руководитель НПЦ болезни двигательного нейрона/БАС, кандидат медицинских наук, врач высшей категории
Аксональная полинейропатия — это одно из самых опасных неврологических заболеваний, сопровождающееся поражением периферической нервной системы. При болезни разрушаются периферические нервные волокна.
Причин возникновение аксональной полинейропатии несколько. Самые распространенные:
- Сахарный диабет нарушает структуру крови, питающей нервы, в свою очередь происходит сбой в обменных процессах.
- Длительный дефицит витаминов В. Именно они максимально важны для правильной работы нервной системы, поэтому долгая нехватка способна привести к аксональной полинейропатии.
- Воздействие токсинов на организм. К ним относят разнообразные отравляющие вещества, например, алкоголь, а также ВИЧ. При отравлении опасными веществами заболевание может развиться уже через несколько дней.
- Наследственный фактор.
- Синдром Гийена-Барре.
- Различные травмы, к которым также относится длительное сдавливание нервов, которое характерно при грыже или остеохондрозе.
Лечение аксональной полинейропатии обязательно должно быть комплексным, иначе нужного эффекта достичь не удастся. Категорически запрещается заниматься самолечением и при возникновении первых же симптомов нужно срочно обратиться к доктору. Врачи Юсуповской больницы подбирают лечение индивидуально для каждого пациента. В зависимости от тяжести патологии и симптоматики назначается комплексное лечение под наблюдением опытных специалистов.

Причины
Самые распространенные причины возникновения аксональной полинейропатии нижних конечностей:
- истощение организма;
- длительный недостаток витаминов группы В;
- недуги, ведущие к дистрофии;
- острые инфекции;
- токсическое поражение ртутью, свинцом, кадмием, угарным газом, спиртными напитками, метиловым спиртом, фосфорорганическими соединениями, медицинскими препаратами, принимаемыми без согласования с врачом;
- болезни сердечно-сосудистой, кроветворной, кровеносной и лимфатической систем;
- эндокринологические патологии, в том числе инсулинозависимость.
Главными факторами, которые провоцируют развитие моторной или сенсомоторной аксональной полинейропатии, являются:
- эндогенная интоксикация при почечной недостаточности;
- аутоиммунные процессы, протекающие в организме;
- амилоидоз;
- вдыхание токсических веществ или паров.
Также болезнь может быть обусловлена наследственностью.
Нехватка в организме витаминов группы В, а в особенности пиридоксина и цианокобаламина, крайне негативно воздействует на проводимость нервных и моторных волокон и может вызывать сенсорную аксональную полинейропатию нижних конечностей. Это же происходит при хронической алкогольной интоксикации, глистной инвазии, заболеваниях желудочно-кишечного тракта, которые ухудшают скорость всасывания.
Токсическое отравление лекарственными препаратами, аминогликозидами, золотыми солями и висмутом занимают большой процент в структуре факторов аксональной невропатии.
У пациентов с сахарным диабетом нарушена функция периферических нервов из-за нейротоксичности кетоновых тел, то есть метаболитов жирных кислот. Происходит это из-за невозможности организма использовать глюкозу как главный источник энергии. Поэтому вместо нее окисляются жиры.
При аутоиммунных заболеваниях, протекающих в организме, иммунная система человека атакует собственные нервные волокна, воспринимая их как источник опасности. Это происходит из-за провокации иммунитета, возникающей при неосторожном приеме иммуностимулирующих медикаментов и нетрадиционных методик лечения. Поэтому у людей, которые склонны к возникновению аутоиммунных заболеваний, пусковыми факторами аксональной полинейропатии являются:
- иммуностимуляторы;
- вакцины;
- аутогемотерапия.
При амилоидозе в организме накапливается такой белок, как амилоид. Именно он нарушает основные функции нервных волокон.
Первые признаки
Заболевание обычно начинает развиваться с поражения толстых или тонких нервных волокон. Зачастую аксональная полинейропатия имеет дистальное симметричное распределение на кисти или стопы. Нейропатия чаще всего сначала поражает нижние конечности, а затем симметрично распространяется вверх по телу. К самым частым первичным симптомам поражения относят:
- мышечную слабость;
- болевой синдром в конечностях;
- жжение;
- ощущение ползания мурашек;
- онемение кожных покровов.
Симптоматика ярче всего проявляется в вечернее и ночное время суток.
Симптомы
Врачи подразделяют хроническое, острое и подострое течение аксональной полинейропатии. Заболевание подразделяется на два вида: первично-аксональный и демиелинизирующий. В ходе течения болезни к ней присовокупляется демиелинизация, а затем и вторично аксональный компонент.
К основным проявлениям недуга относятся:
- вялость в мышцах ног или рук;
- спастический паралич конечностей;
- чувство подергивания в мышечных волокнах;
- головокружение при резкой перемене положения тела;
- отек конечностей;
- жжение;
- покалывание;
- ощущение ползания мурашек;
- снижение чувствительности кожных покровов к высокой или низкой температуре, боли и касаниям;
- нарушение ясности речи;
- проблемы с координацией.
Вегетативными признаками сенсомоторной полинейропатии асконального типа считаются следующие симптомы:
- учащенный или, напротив, замедленный сердечный ритм;
- неумеренное потоотделение;
- чрезмерная сухость кожи;
- изменение цвета кожных покровов;
- нарушение эякуляции;
- эректильная дисфункция;
- проблемы с мочеиспусканием;
- сбой двигательных функций желудочно-кишечного тракта;
- повышенное слюнотечение или, наоборот, сухость во рту;
- расстройство аккомодации глаза.
Заболевание проявляется в нарушениях функций поврежденных нервов. Именно периферические нервные волокна отвечают за двигательные функции мышечной ткани, чувствительность, а также оказывают вегетативное воздействие, то есть регулируют сосудистый тонус.
Для нарушения функции проводимости нервов характерны расстройства чувствительности, например:
- чувство ползания мурашек;
- гиперестезия, то есть увеличение чувствительности кожи к внешним раздражителям;
- гипестезия, то есть уменьшение чувствительности;
- отсутствие ощущения собственных конечностей.
Когда поражены вегетативные волокна, то из-под контроля выходит регуляция сосудистого тонуса. При аксонально-демиелинизирующей полинейропатии наступает сдавление капилляров, из-за чего ткани отекают. Нижние, а затем и верхние конечности из-за скапливания в них жидкости существенно увеличиваются в размерах. Так как при полинейропатии нижних конечностей основное количество крови накапливается именно в пораженных областях тела, то у пациента возникает стойкое головокружение при принятии вертикального положения. Из-за того, что пропадает трофическая функция, могут возникнуть эрозивно-язвенные поражения нижних конечностей.
Аксональная моторная полинейропатия проявляется в двигательных нарушениях верхних и нижних конечностей. Когда моторные волокна, отвечающие за движения рук и ног, повреждены, то наступает полный или частичный паралич мышц. Обездвиживание может проявляться совершенно нетипично — может ощущаться как скованность мышечных волокон, так и чрезмерная их расслабленность. При средней степени поражения ослаблен мышечный тонус.
В ходе течения заболевания могут быть усилены или ослаблены сухожильные и надкостничные рефлексы. В редких случаях доктор-невролог их не наблюдает. При болезни часто могут быть поражены черепные нервы, которые проявляются следующими нарушениями:
- глухотой;
- онемением подъязычных мышц и мускулатуры языка;
- невозможностью проглотить еду или жидкость из-за проблем с глотательным рефлексом.
Когда поражен тройничный, лицевой или глазодвигательный нерв, изменяется чувствительность кожных покровов, развиваются параличи, возникает асимметрия лица и подергивание мышц. Иногда при диагностированной аксонально-демиелинизирующей полинейропатии поражения верхних или нижних конечностей могут быть асимметричными. Такое случается при множественной мононейропатии, когда коленные, ахилловы и карпорадиальные рефлексы несимметричны.
Диагностика
Главной методикой исследования, которая позволяет обнаружить локализацию патологического процесса и степень пораженности нервов, является электронейромиография.
Чтобы определить причину заболевания, врачи назначают следующие анализы:
- определение уровня сахара в плазме крови;
- токсикологические тесты;
- полный анализ мочи и крови;
- выявление уровня холестерина в организме.
Нарушение нервных функций устанавливается при помощи определения температурной, вибрационной и тактильной чувствительности.
При первичном осмотре применяется зрительная методика исследования. То есть врач, к которому обратился с жалобами пострадавший, осматривает и анализирует такие внешние симптомы, как:
- уровень давления крови в верхних и нижних конечностях;
- чувствительность кожных покровов к прикосновениям и температуре;
- наличие всех необходимых рефлексов;
- диагностика отечности;
- изучение внешнего состояния кожи.
Выявить аксональную полинейропатию можно при помощи следующих инструментальных исследований:
- магнитная резонансная томография;
- биопсия нервных волокон;
- электронейромиография.
Лечение аксональной полинейропатии
Лечение аксональной полинейропатии должно быть комплексным и направленным на причину развития заболевания, его механизмы и симптоматику. Гарантией эффективной терапии является своевременное выявление болезни и лечение, которое сопровождается абсолютным отказом от сигарет, алкоголя и наркотических веществ, ведением здорового образа жизни и соблюдением всех рекомендаций врача. В первую очередь проводятся следующие терапевтические мероприятия:
- избавление от токсического воздействия на организм, если оно присутствует;
- антиоксидантная терапия;
- прием препаратов, которые воздействуют на тонус кровеносных сосудов;
- восполнение дефицита витаминов;
- регулярный контроль концентрации глюкозы в плазме крови.
Отдельное внимание уделяется лечению, направленному на купирование острого болевого синдрома.
Если присутствуют периферические парезы, то есть существенное снижение мышечной силы с многократным уменьшением амплитуды движений, то в обязательном порядке показана лечебная физкультура и специальные физические упражнения, направленные на возвращение тонуса мышечным тканям и предотвращение образования различных контрактур. Особенно важна регулярная психологическая поддержка, которая не дает пациенту впасть в депрессию, сопровождающуюся расстройством сна и чрезмерной нервной возбудимостью.
Лечение аксональной полинейропатии — это продолжительный процесс, так как нервные волокна восстанавливаются долго. Поэтому не стоит ожидать моментального выздоровления и возвращения к привычному образу жизни. Медикаментозная терапия включает такие препараты, как:
- обезболивающее;
- глюкокортикоиды;
- витамины группы В;
- антиоксиданты;
- сосудорасширяющие;
- средства, ускоряющие метаболизм и улучшающие микроциркуляцию крови.
Терапия лекарственными препаратами направлена на восстановление функций нервов, улучшение проводимости нервных волокон и скорости передачи сигналов центральной нервной системе.
Лечение следует проводить длительными курсами, которые не стоит прерывать, хоть и эффект от них проявляется не сразу. Чтобы устранить болевые ощущения и расстройство сна, назначают следующие медикаменты:
- антидепрессанты;
- противосудорожные;
- препараты, купирующие аритмию;
- обезболивающие.
Для избавления от боли используют нестероидные противовоспалительные препараты. Но стоит помнить, что применять их можно только короткий промежуток времени, так как длительное употребление может привести к повреждению слизистой оболочки желудочно-кишечного тракта.
К физиотерапевтическим методам лечения аксональной полинейропатии относятся:
- терапия магнитными волнами;
- грязелечение;
- электростимуляция;
- иглоукалывание;
- лечебный массаж;
- физкультура;
- ультрафонофорез;
- гальванотерапия.
Именно лечебная физкультура позволяет сохранить работоспособность мышечных тканей и поддерживать конечности в нужном положении. Регулярные занятия спортом вернут мышцам тонус, гибкость и увеличат амплитуду движений до нормальной.
Прогноз
Если заболевание обнаружено на ранней стадии и комплексно лечится квалифицированными специалистами, то прогноз для жизни и здоровья пациента более чем благоприятный. Стоит вести правильный образ жизни, рацион должен быть богат витаминами и минералами, необходимыми для правильного функционирования организма.
Если долгое время игнорировать болезнь и не предпринимать никаких действий, результат будет плачевным вплоть до полного паралича.
Профилактика
Пациент в обязательном порядке должен совершать профилактические мероприятия, которые помогут избежать рецидива или возникновения опасного заболевания. Они включают в себя обогащение рациона витаминами, регулярный контроль уровня сахара в крови, полный отказ от табакокурения, наркотических веществ и алкогольных напитков.
В целях профилактики болезни рекомендуется:
- носить удобную обувь, которая не пережимает стопу, ухудшая кровоток;
- регулярно осматривать обувь, чтобы избежать образования грибка;
- исключить пешие прогулки на длительные расстояния;
- не стоять долгое время на одном месте;
- мыть ноги прохладной водой или делать контрастные ванночки, что помогает улучшить циркуляцию крови в организме.
Пострадавшим в стадии ремиссии категорически запрещается принимать лекарственные препараты без согласования с лечащим врачом. Важно своевременно лечить воспалительные заболевания, соблюдать меры предосторожности при работе с токсическими веществами, которые оказывают пагубное воздействие на организм, регулярно выполнять лечебные физические упражнения.
 Диффузное аксональное повреждение головного мозга (далее ДАП) – это черепно-мозговая травма, результатом которой является разрыв или повреждение аксонов (отростков нервных клеток), сообщающих нервные импульсы клеткам ЦНС, органам и тканям.
Диффузное аксональное повреждение головного мозга (далее ДАП) – это черепно-мозговая травма, результатом которой является разрыв или повреждение аксонов (отростков нервных клеток), сообщающих нервные импульсы клеткам ЦНС, органам и тканям.
ДАП, зачастую, ведет к коме, в результате которой человек может перейти в вегетативное состояние.
Диффузия головного мозга чаще происходит с молодыми людьми, которые попадают в дорожно-транспортные происшествия, становятся жертвами драк и избиений с повреждением головного мозга, а также детьми, у которых и кома намного глубже, и неврологические нарушения грубее.
Вариант этой ЧМТ был впервые описан в 1956 за рубежом, а само название появилось в 1982 году. Состояние при ДАП тяжелое, протекающее в длительной коме, которая сразу возникает после травмы и характеризуется длительным течением.
Аксоны, поврежденные или разорванные в результате ЧМТ, и мелкие кровоизлияния равномерно распределяются по церебральным структурам, при этом нарушается иннервация всех зависимых органов. Самые распространенные места повреждений:
- ствол мозга;
- белое вещество;
- мозольное тело;
- волокна перивентрикулярные.
Диффузная травма головного мозга – это всегда серьезнейшее состояние с перспективой перехода в вегетативное состояние, нередок и смертельный исход.
Причины и морфология ДАП
Самые распространенные причины ЧМТ при ДАП:
- сильный ушиб о лобовые стекла в ДТП;
- падение;
- удар тяжелым предметом;
- синдром сотрясения у детей, при котором мозг подвергается сильному ушибу впоследствии резкой тряски, а также избиения, падения.
Синдром ДАП является результатом ушибов, вызванных угловым ускорением головы. При этом может и не быть прямого столкновения с объектом травмы.
Вследствие этого у некоторых пациентов не наблюдается переломов черепа и других видимых ранений, что несколько затрудняет диагностику. Статистика говорит о том, что именно повреждение по косой ведет к повреждению аксонов и возникновению ДАП.
Морфологически для этой травмы характерны три следующих очага повреждения:
- головной мозг;
- мозольное тело;
- стволовой мозг и диффузно распределенные разрывы.
Первые два очаговых признака – макроскопические, которые располагаются в виде гематомы до пяти мм, выглядящие как надорванная ткань с окровавленными краями. Через несколько дней после ЧМТ, очаг пигментируется и затем рубцуется. Рана в мозолистом теле может разрешиться образованием сосудистой кисты.

Аксональные нарушения при травме
Особенности клинической картины
Кома при диффузном повреждении головного мозга продолжается до трех недель и проявляется следующими симптомами:
- рефлекс зрачка нарушается;

- происходит паралич взгляда;
- дыхательный ритм меняется;
- повышается тонус мышц;
- появляется парез ног и рук;
- диагностируется гипертония;
- появляется фебрильная или субфебрильная температура и другие вегетативные расстройства.

Когда пациент с ДАП выходит из комы, он оказывается в вегетативном состоянии со следующими симптомами:
- глаза открываются на раздражители или самостоятельно;
- взгляд не сконцентрирован и не следует за движущимися предметами.
Вегетативное состояние, сопровождающееся нарушением рефлексов и симптомами разъединения функций мозговых полушарий, может продолжаться, в среднем, от нескольких дней до нескольких лет. Чем больше нахождение в нем, тем скорее появляются такие признаки полиневропатии, как:
- ослабление кистей рук;
- хаотическое движение мышц;
- расстройства нейротрофики;
- учащение пульса;
- отеки;
- тахипноэ и др.
После выхода из вегетативного состояния личность выпадает. Основные признаки нарушений после выхода:
- экстрапирамидные расстройства;
- расстройства психики, проявляющиеся отсутствием интереса к окружающей пациента действительности, агрессия, амнезия, слабоумие.
Степени поражения
Диффузное аксональное повреждение головного мозга бывает трех степеней тяжести:
- легкой – продолжительность комы от 6 до 24 часов, ЧМТ незначительна;
- средней– кома больше 24 часов, но черепно-мозговая травма умеренная;
- тяжелой – длительная кома, поражения значительные, происходит сдавливание мозга.
Тяжелая степень характеризуется массовым повреждением аксонов, которое приводят к кровоизлияниям в мозг. ДАП при этом может приводить к коме, продолжающейся годами и летальному исходу.
Начальное состояние мозга после выхода из такой комы восстановить не представляется возможным, единицы вернулись к более-менее нормальной жизни после таких повреждений.
Постановка диагноза
Диагноз диффузное поражение головного мозга ставится после компьютерной томографии, результаты которой в остром периоде отличаются увеличением объема мозговых полушарий, уменьшением или сдавливанием боковых расстояний и основания мозга. В белом веществе, стволе и мозолистом теле находятся мелкие кровоизлияния.
При обследовании наблюдается молниеносное развитие признаков ДАП и дегенерации.
ЭЭГ при синдроме ДАП выявляет изменения в подкорке и стволе мозга, диэнцефальный синдром. В анализе крови отмечается резкое повышение серотонина, значительное уменьшение дофамина и скачок адреналина, что предполагает терапию, направленную на снижение симпатико-адреналиновых симптомов.
В результате КТ определяют повышено внутричерепное давление или, наоборот, понижено либо отсутствует. В таком случае подключают датчик. Если на КТ отток ликвора в норме, то и внутричерепное давление будет в норме.

Поддержка состояния пострадавшего
После диффузной травмы головного мозга нередко диагностируются субдуральные ликворные скопления над большими полушариями мозга, которые в дальнейшем рассасываются, удалять хирургически их не надо.
Аксональное повреждение головного мозга чаще всего лечится консервативно. Нейрохирургическая операция проводится при сочетании разрывов и повреждений аксонов с очаговыми повреждениями, усиливающими сдавливание и провоцирующими гидроцефальный синдром.
В коме пациента подключают к ИВЛ, кормят парентерально и вводят следующие лекарства:
- для установления правильного кислотно-щелочного и водно-электролитного баланса;
- ноотропные и вазоактивные;
- устраняющие гипертензию или гипотонию;
- антибиотики для исключения сопутствующих инфекций.
Для возобновления психэмоциональной сферы вводят прием психостимуляторов.
После выхода из комы:
- вводят ноотропы и сосудистые лекарства для нормализации и улучшения состояния ЦНС, ноотропы также важны для последующей реабилитации;
- назначают препараты для улучшения метаболизма и биостимуляторы;
- проводят лечебную физкультуру для профилактики парезов;
- пациент занимается с логопедом.
Гормональные препараты при ДАП не назначают по ненадобности. После операции, если она все же состоялась (произошло сдавление головного мозга при сопутствующих травмах), вводят препараты, препятствующие образованию отеков, сосудистые средства, ноотропы, антихолинэстеразные, психотропные (во избежание развития агрессии и депрессии) и нейромедиаторы.
В период восстановления проводится такая же терапия, как и после выхода из комы.
Исход тяжелой травмы и ее последствия
Прогноз и последствия диффузного аксонального повреждения зависит от степени поражения аксонов головного мозга и тяжести вторичных признаков, таких как повышенное внутричерепное давление, гипергидроз, разбухание мозговых оболочек, психические нарушения, развитие слабоумия и т. д.
Исход также зависит от того как помогают лечебные методы, направленные на ликвидацию последствий ДАП – вторичные повреждения и осложнения.
Прогноз предполагает, что чем больше человек находился в коматозном состоянии, тем более риск развития неблагоприятных поражений, вплоть до летального исхода. Шансов на восстановление будет также минимум.
 Но, нужно сказать, что иногда восстановиться полностью или почти полностью, вернуть психические функции, вернуться к нормальной деятельности, убрать все неврологические нарушения можно, даже если человек находился в коме третьей степени (тяжелой), а после продолжительное время пребывал в вегетативном состоянии. Тенденция к самовосстановлению всегда присутствует у мозга, известны и более тяжелые нарушения, при которых он восстанавливался.
Но, нужно сказать, что иногда восстановиться полностью или почти полностью, вернуть психические функции, вернуться к нормальной деятельности, убрать все неврологические нарушения можно, даже если человек находился в коме третьей степени (тяжелой), а после продолжительное время пребывал в вегетативном состоянии. Тенденция к самовосстановлению всегда присутствует у мозга, известны и более тяжелые нарушения, при которых он восстанавливался.
Но, к сожалению, чаще у выживших людей последующее течение синдрома ДАП может идти по двум сценариям:
- выход из коматозного состояния;
- переход в вегетативное состояние.
При первом варианте глаза больного открываются, и происходит слежение за предметами и фиксация взгляда на объекте. Это может иметь как спонтанный выход, так и направляемый организованными раздражителями, звуком и болевыми манипуляциями.
Затем пациент восстанавливает сознание, выполняет обращенные к нему просьбы, словесный багаж расширяется, он начинает общаться. Неврологические патологии при этом медленно регрессируют.
У больных, которые вышли из вегетативного состояния, развиваются экстрапирамидальные симптомы, сопровождающиеся психическими нарушениями (слабоумие, лабильность настроения, аспонтанность, спутанность сознания). При втором варианте летальный исход через определенное время неизбежен из-за истощения нейромедиаторов и соматических осложнений.
Современные исследования подтверждают регенерацию аксонов у детей и молодых людей, у которых мозг еще не завершил формирование. Происходит восстановление неврологических и психических процессов. При продолжительной коме оно проблематично, инвалидизация гарантирована.
Российский государственный медицинский университет;
НИИ цереброваскулярной патологии и инсульта, Москва

Аксональные полинейропатии: патогенез и лечение
Как цитировать:
Ковражкина Е.А. Аксональные полинейропатии: патогенез и лечение. Журнал неврологии и психиатрии им. С.С. Корсакова.
2013;113(6):22‑25.
Kovrazhkina EA. Axonal polyneuropathies: pathogenesis and treatment. Zhurnal Nevrologii i Psikhiatrii imeni S.S. Korsakova. 2013;113(6):22‑25. (In Russ.)
Полинейропатия (ПНП) — заболевание периферической нервной системы, развивающееся в результате диффузного поражения периферических нервов — их аксонов (аксональные полинейропатии), миелиновой оболочки (демиелинизирующие полинейропатии) либо тел нейронов (нейронопатии) [9]. В основе патогенеза полинейропатий аксонального типа лежит генерализованное повреждение осевых цилиндров периферических нервов.
Патогенез. Аксональная дегенерация (аксонопатия) — результат нарушения метаболизма нейрона вследствие недостаточной выработки энергии в митохондриях и/или нарушения аксонального транспорта. Миелиновая оболочка при аксонопатиях страдает вторично (вторичная демилинизация). Миелин может повреждаться в результате ишемии нервов (поражения vasa nevrorum), отложения токсичных для нерва веществ или иммунных комплексов в эндоневрии (что характерно, например, для сахарного диабета, особенно 1-го типа — с высокой гипергликемией и аутоиммунными нарушениями) [4]. Соответственно, среди причин аксонопатии периферических нервов указывают нарушения метаболизма, токсические влияния, ишемию нервных стволов, наследственную предрасположенность и аутоиммунные механизмы [5]. По этиологическому фактору подавляющее большинство аксональных ПНП составляют метаболические и токсические, среди которых первое место занимают диабетическая (до 50% среди пациентов с сахарным диабетом и до 90% поражений нервной системы при диабете [9, 12]) и алкогольная (до 50% среди больных алкоголизмом [2, 9]).
Причиной токсико-метаболических ПНП является экзо- и эндогенные интоксикации. Поступающие извне вещества или собственные его метаболиты, токсичные для периферических нервов, вызывают их повреждение. Тяжесть этого повреждения зависит от степени токсичности данного агента (выделяют острые метаболические ПНП, развивающиеся на фоне тяжелой общей интоксикации, например, ПНП при быстро нарастающих печеночной недостаточности или уремии, при отравлении фосфоорганическими веществами, мышьяком, свинцом, как нежелательный эффект лечения препаратами лития, цитостатиками), длительности его воздействия, собственных генетических особенностей метаболизма нервной ткани, немаловажную роль играет также аутоиммунный фактор [13].
Большинство часто встречающихся в клинике метаболических ПНП являются итогом длительного воздействия эндо- и экзогенного токсического агента (диабетическая, печеночная, уремическая, алкогольная, профессиональная, лекарственная). Результатом такой интоксикации является повреждение осевого цилиндра аксона. Клинически это проявляется не только нарушениями чувствительности и мышечной слабостью (что свойственно и для демиелинизирующих ПНП), но и мышечными гипотрофиями, выраженными трофическими нарушениями. Все эти признаки свидетельствуют о длительном страдании аксонов периферических нервов. При электронейромиографии (ЭНМГ) выявляется снижение (иногда — вплоть до полного отсутствия) амплитуды сенсорных потенциалов и М-ответов периферических нервов. У пациентов с ПНП аксонального типа зачастую в достаточной степени сохранены двигательные функции (отсутствуют выраженные парезы, больные сохраняют способность ходить, часто без дополнительной опоры), тогда как инвалидизирующими являются чувствительные (боли и парестезии) и трофические нарушения. Так, аксональная сенсомоторная ПНП является одним из важных патогенетических факторов развития синдрома диабетической стопы [3]. Аксональное повреждение нервов развивается медленно, исподволь, но при правильном лечении потенциально обратимо.
При массивном воздействии токсичного для периферической нервной системы агента, участии ишемического компонента (за счет страдания vasa nevrorum), аутоиммунных влияний развиваются ПНП аксонально-демиелинизирующего типа такие, например, как уремическая, свинцовая, амиодароновая, вызываемые воздействиями высокотоксичных для нервов веществами [9]. При наиболее распространенной диабетической ПНП, демиелинизирующий компонент максимально представлен при инсулинпотребном сахарном диабете (характеризующимся более высокой гипергликемией), а выраженность демиелинизации периферических нервов нарастает при резких повышениях уровня глюкозы крови [4, 7]. На представленность демиелинизирующего компонента при метаболических ПНП влияют наследственные факторы (генетически обусловленная миелинопатия, которая протекала бы субклинически без дополнительного воздействия токсического агента), аутоиммунное повреждение (например, миелинопатия более выражена при сахарном диабете 1-го типа, при котором иммунокомпетентные клетки повреждают ткань поджелудочной железы) [4, 14].
ПНП аксонально-демиелинизирующего типа протекают более тяжело, с выраженными парезами, сенситивной атаксией, нейропатическим болевым синдромом, но нередко, при своевременном устранении действия токсического фактора, быстро регрессируют. При очень тяжелом и упорном течении заболевания, учитывая роль аутоиммунного механизма в развитии данного типа метаболических ПНП, необходимо кратковременное назначение иммуномодулирующей терапии (чаще глюкокортикоидов, иногда цитостатиков) [9, 11].
Клиническое течение ПНП. Как уже было сказано, при длительном воздействии умеренно токсичного для нервов агента аксонопатия развивается медленно, что не всегда заметно. Преимущественное поражение бедно миелинизированых вегетативных и сенсорных волокон вызывает парестезии, повышенную холодовую чувствительность кистей и стоп, негрубые трофические нарушения. При ЭНМГ не всегда удается выявить характерные признаки сенсорной и моторной аксонопатии — снижение амплитуд сенсорных потенциалов и М-ответов — если исследуются крупные богато миелинизированные нервы, аксоны которых страдают позднее. Быстрее выявляются признаки вторичной миелинопатии, как клинические — присоединение нейропатических болей, парезов, так и миографические — снижение скорости распространения возбуждения (СРВ) по нервам.
При условии адекватной терапевтической стратегии — устранения действия повреждающего периферические нервы фактора, назначение препаратов, влияющих на метаболизм нервной ткани, — наступает ремиелинизация, что отражается и на ЭНМГ в виде нарастания СРВ, и клинически — в виде регресса парезов и нейропатических болей, улучшения чувствительности. Однако, если полинейропатия развивалась в условиях длительного действия токсического агента, поражение аксонов периферических нервов, снижение амплитуд сенсорных потенциалов и М-ответов на ЭНМГ сохраняется, поскольку осевые цилиндры нервов обладают гораздо меньшей способностью к регенерации, чем миелин.
Так, в проведенном исследовании [8], посвященном лечению алкогольной ПНП, ведущим ЭНМГ-признаком поражения периферических нервов у большинства пациентов имелось снижение СРВ. В условиях элиминации токсического фактора (в данном случае приема алкоголя) и лечения препаратом α-липоевой кислоты (берлитион в суточной дозе 600 мг), у пациентов быстро, в течение 1-го месяца, регрессировали ЭНМГ и клинические признаки миелинопатии — уменьшалась выраженность парезов и болевого синдрома (в баллах по визуальной аналоговой шкале с 5,2±1,0 до 2,6±0,5) достоверно (р<0,05) нарастала СРВ (по моторным волокнам n. tibialis с 36,93±1,12 до 42,22±0,8 м/с, по сенсорным волокнам n. suralis c 32,45±0,70 до 38,24±0,50 м/с), но признаки аксонопатии (снижение амплитуд сенсорных и моторных ответов периферических нервов, сенсорные выпадения по полиневритическому типу, трофические нарушения) сохранялись и после 6 нед лечения.
Таким образом, можно представить следующую схему развития и течения метаболических ПНП (см. таблицу): высокотоксичные, высококонцентрированные токсические агенты приводят к развитию полинейропатии аксонально-демиелинизирующего типа (в развитии которых также значительную роль играют наследственный, сосудистый, аутоиммунный факторы), длительно действующие эндо- и экзогенные яды приводят преимущественно к страданию аксонов с умеренно выраженным вторичным поражением миелина (так как постоянно идет процесс ремиелинизации) [7].
Лечение. Аксональное повреждение нервов как первичного, так и вторичного характера, обратимо за счет регенерации поврежденных аксонов и концевого спрутинга сохранившихся аксонов, однако этот процесс протекает медленно (месяцы), часто аксональная регенерация бывает неполной. Для регресса аксонопатии периферических нервов необходимо нивелировать воздействие токсического агента (коррекция метаболических и алиментарных нарушений, детоксикация, влияние на аутоиммунные механизмы), большое значение имеет также патогенетическая терапия, направленная на восстановление нарушенного метаболизма аксонов (митохондриальные нарушения, повреждения, вызванные окислительным стрессом).
Таким образом, в патогенетическом лечении аксональных ПНП, в зависимости от их нозологического и клинико-электрофизиологического варианта, должны присутствовать детоксикация и коррекция метаболических и алиментарных нарушений; сосудистая терапия (дезагреганты, венотоники, пентоксифиллин, вазопростан); препараты, воздействующие на универсальные механизмы поражения аксонов (витамины и витаминоподобные препараты); при необходимости — воздействие на аутоиммунные компоненты (глюкокортикоиды, плазмаферез). В некоторых случаях, когда представленность миелинопатии в структуре поражения периферических нервов велика и трудно отличить токсико-метаболическую полинейропатию от воспалительной (например, синдром Гийена-Барре у пациента с алкоголизмом или дебют хронической воспалительной демиелинизирующей ПНП у больного с сахарным диабетом) необходимо проводить пробную иммуномодулирующую терапию, ее быстрая эффективность позволит высказаться в пользу превалирования у пациента аутоиммуного механизма поражения периферических нервов [7, 11].
В лечении всех метаболических ПНП необходимо устранить (по возможности) поражающий периферические нервы яд и использовать препараты, улучшающие метаболизм нервной ткани периферических нервов. Последние необходимо применять в течение длительного времени, так как первым эффектом данных препаратов будет ускорение ремиелинизации, но для восстановления самих аксонов — а данные ПНП являются аксональными — требуется значительно большее количество времени.
α-липоевая (тиоктовая) кислота — витаминоподобное вещество, эндогенно образующееся в организме, как кофермент участвует в окислительном декарбоксилировании α-кетокислот. Основная функция эндогенной липоевой кислоты в организме — участие в аэробном метаболизме продукта гликолиза пирувата. Тиоктовая кислота является коферментом в окислительном декарбоксилировании пировиноградной кислоты до ацетил-КоА и α-кетоглутаровой до сукцинил-КоА в цикле Кребса. Облегчая превращение молочной кислоты в пировиноградную с последующим декарбоксилированием последней, α-липоевая способствует ликвидации метаболического ацидоза [6]. Тиоктовая кислота обладает сложным комплексным действием: гипогликемическим, липотропным, гепатопротекторным, антиатеросклеротическим, является мощным антиоксидантом. Липоевая кислота может существовать в окисленной (-S-S-) и восстановленной (SH-)-формах, благодаря чему реализуются ее коферментные и антиоксидантные функции. Восстановленная форма, дигидролипоевая кислота, служит донором электронов для восстановления других антиоксидантов (витамины С, Е и глутатион), осуществляет рецикл витамина Е при его истощении. Дигидролипоат повышает интра- и экстрацеллюлярный уровни глутатиона — эндогенного антиоксиданта.
Эффект экзогенно вводимой α-липоевой кислоты в отношении полиневритического синдрома впервые обнаружен при сахарном диабете. Обусловленная сахарным диабетом гипергликемия приводит к отложению глюкозы на матричных протеинах кровеносных сосудов и образованию конечных продуктов прогрессирующего гликозилирования, в результате чего уменьшается эндоневральный кровоток, возникает эндоневральная ишемия. α-липоевая кислота приводит к снижению уровня глюкозы в крови и повышению содержания гликогена в печени, обладает гипогликемическим действием. На фоне воздействия препарата уменьшается выраженность сенсорных симптомов полинейропатии — боли, жжения, ощущения онемения и «ползания мурашек» в конечностях.
Применение α-липоевой кислоты оказывает положительное влияние на универсальные механизмы аксонального повреждения, такие как повреждающее действие окислительного стресса и митохондриальная дисфункция — за счет антиоксидантного действия, повышения содержания глутатиона. Энергокорригирующее действие α-липоевой кислоты, тропное именно к аксонам нервов, способствует, в конечном итоге, быстрейшей регенерации аксонов [18].
Препарат не только редуцирует проявления окислительного стресса, но и оказывает влияние на сосудистый компонент поражения периферических нервов, нормализуя эндоневральный кровоток (что имеет большое значения, например, при дибетической микроангиопатии) [16, 17]. Комплексный механизм действия α-липоевой кислоты объясняет ее эффективность в отношении всех аксональных ПНП, патогенез которых связан с токсико-дисметаболическим и сосудистым факторами. Так, эффективность тиоктовой кислоты показана при уремической и алкогольной ПНП, при поражении периферических нервов, индуцированном цитостатиками [15]. Для регенерации аксонов периферических нервов на фоне токсико-метаболических влияний важны также детоксикационный и гепатопротекторный эффекты α-липоевой кислоты. Положительный эффект препарата отмечен в отношении заболеваний печени, печеночной комы, некоторых интоксикаций, в том числе алкогольной [6, 10].
При значительно выраженной аксонопатии, учитывая медленную скорость регенерации аксонов, необходимо длительное применение достаточно высоких доз α-липоевой кислоты. Обычно ее суточная доза составляет 600 мг. Предпочтительнее начинать лечение с внутривенного капельного введения препарата — 600 мг (24 мл раствора) в разведении на 200 мл физиологического раствора, длительность инфузии составляет от 2 до 4 нед в зависимости от тяжести ПНП. В особо тяжелых случаях препарат вводят внутривенно капельно в дозе 1200 мг в сутки. После переходят на пероральный прием α-липоевой кислоты — в таблетках по 600 мг не менее 2 месяцев [1].
В соответствии с такой клинической потребностью — необходимостью длительного приема достаточно высоких доз α-липоевой кислоты, учитывая потенциально обратимый, но медленный характер аксональной регенерации — разработаны и фармацевтические формы препарата. Например, берлитион 300 и берлитион 600, производимые в виде раствора для внутривенных вливаний по 12 и 24 мл соответственно (для начального этапа лечения ПНП), так и таблеток по 300 и 600 мг (для продолжения терапии).
Соблюдение всех условий патогенетического лечения ПНП — нормализация гликемии, прекращение поступления экзогенного яда, детоксикация, коррекция аутоиммунных нарушений, длительное применение препаратов, нормализующих аксональный метаболизм, способствует постепенной регенерации аксонов, уменьшению выраженности чувствительных и двигательных нарушений, болевого синдрома, парестезий, парезов. Для уточнения динамики различных видов метаболических ПНП на фоне подобного терапевтического подхода необходимы дальнейшие исследования.
| Axon | |
|---|---|
An axon of a multipolar neuron |
|
| Identifiers | |
| MeSH | D001369 |
| FMA | 67308 |
| Anatomical terminology
[edit on Wikidata] |
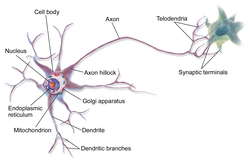
An axon (from Greek ἄξων áxōn, axis), or nerve fiber (or nerve fibre: see spelling differences), is a long, slender projection of a nerve cell, or neuron, in vertebrates, that typically conducts electrical impulses known as action potentials away from the nerve cell body. The function of the axon is to transmit information to different neurons, muscles, and glands. In certain sensory neurons (pseudounipolar neurons), such as those for touch and warmth, the axons are called afferent nerve fibers and the electrical impulse travels along these from the periphery to the cell body and from the cell body to the spinal cord along another branch of the same axon. Axon dysfunction can be the cause of many inherited and acquired neurological disorders that affect both the peripheral and central neurons. Nerve fibers are classed into three types – group A nerve fibers, group B nerve fibers, and group C nerve fibers. Groups A and B are myelinated, and group C are unmyelinated. These groups include both sensory fibers and motor fibers. Another classification groups only the sensory fibers as Type I, Type II, Type III, and Type IV.
An axon is one of two types of cytoplasmic protrusions from the cell body of a neuron; the other type is a dendrite. Axons are distinguished from dendrites by several features, including shape (dendrites often taper while axons usually maintain a constant radius), length (dendrites are restricted to a small region around the cell body while axons can be much longer), and function (dendrites receive signals whereas axons transmit them). Some types of neurons have no axon and transmit signals from their dendrites. In some species, axons can emanate from dendrites known as axon-carrying dendrites.[1] No neuron ever has more than one axon; however in invertebrates such as insects or leeches the axon sometimes consists of several regions that function more or less independently of each other.[2]
Axons are covered by a membrane known as an axolemma; the cytoplasm of an axon is called axoplasm. Most axons branch, in some cases very profusely. The end branches of an axon are called telodendria. The swollen end of a telodendron is known as the axon terminal which joins the dendron or cell body of another neuron forming a synaptic connection. Axons make contact with other cells – usually other neurons but sometimes muscle or gland cells – at junctions called synapses. In some circumstances, the axon of one neuron may form a synapse with the dendrites of the same neuron, resulting in an autapse. At a synapse, the membrane of the axon closely adjoins the membrane of the target cell, and special molecular structures serve to transmit electrical or electrochemical signals across the gap. Some synaptic junctions appear along the length of an axon as it extends; these are called en passant («in passing») synapses and can be in the hundreds or even the thousands along one axon.[3] Other synapses appear as terminals at the ends of axonal branches.
A single axon, with all its branches taken together, can innervate multiple parts of the brain and generate thousands of synaptic terminals. A bundle of axons make a nerve tract in the central nervous system,[4] and a fascicle in the peripheral nervous system. In placental mammals the largest white matter tract in the brain is the corpus callosum, formed of some 200 million axons in the human brain.[4]
Anatomy[edit]
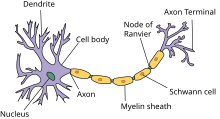
A typical myelinated axon

Axons are the primary transmission lines of the nervous system, and as bundles they form nerves. Some axons can extend up to one meter or more while others extend as little as one millimeter. The longest axons in the human body are those of the sciatic nerve, which run from the base of the spinal cord to the big toe of each foot. The diameter of axons is also variable. Most individual axons are microscopic in diameter (typically about one micrometer (µm) across). The largest mammalian axons can reach a diameter of up to 20 µm. The squid giant axon, which is specialized to conduct signals very rapidly, is close to 1 millimeter in diameter, the size of a small pencil lead. The numbers of axonal telodendria (the branching structures at the end of the axon) can also differ from one nerve fiber to the next. Axons in the central nervous system (CNS) typically show multiple telodendria, with many synaptic end points. In comparison, the cerebellar granule cell axon is characterized by a single T-shaped branch node from which two parallel fibers extend. Elaborate branching allows for the simultaneous transmission of messages to a large number of target neurons within a single region of the brain.
There are two types of axons in the nervous system: myelinated and unmyelinated axons.[5] Myelin is a layer of a fatty insulating substance, which is formed by two types of glial cells: Schwann cells and oligodendrocytes. In the peripheral nervous system Schwann cells form the myelin sheath of a myelinated axon. Oligodendrocytes form the insulating myelin in the CNS. Along myelinated nerve fibers, gaps in the myelin sheath known as nodes of Ranvier occur at evenly spaced intervals. The myelination enables an especially rapid mode of electrical impulse propagation called saltatory conduction.
The myelinated axons from the cortical neurons form the bulk of the neural tissue called white matter in the brain. The myelin gives the white appearance to the tissue in contrast to the grey matter of the cerebral cortex which contains the neuronal cell bodies. A similar arrangement is seen in the cerebellum. Bundles of myelinated axons make up the nerve tracts in the CNS. Where these tracts cross the midline of the brain to connect opposite regions they are called commissures. The largest of these is the corpus callosum that connects the two cerebral hemispheres, and this has around 20 million axons.[4]
The structure of a neuron is seen to consist of two separate functional regions, or compartments – the cell body together with the dendrites as one region, and the axonal region as the other.
Axonal region[edit]
The axonal region or compartment, includes the axon hillock, the initial segment, the rest of the axon, and the axon telodendria, and axon terminals. It also includes the myelin sheath. The Nissl bodies that produce the neuronal proteins are absent in the axonal region.[3] Proteins needed for the growth of the axon, and the removal of waste materials, need a framework for transport. This axonal transport is provided for in the axoplasm by arrangements of microtubules and intermediate filaments known as neurofilaments.
Axon hillock[edit]

Detail showing microtubules at axon hillock and initial segment.
The axon hillock is the area formed from the cell body of the neuron as it extends to become the axon. It precedes the initial segment. The received action potentials that are summed in the neuron are transmitted to the axon hillock for the generation of an action potential from the initial segment.
Axonal initial segment[edit]
The axonal initial segment (AIS) is a structurally and functionally separate microdomain of the axon.[6][7] One function of the initial segment is to separate the main part of an axon from the rest of the neuron; another function is to help initiate action potentials.[8] Both of these functions support neuron cell polarity, in which dendrites (and, in some cases the soma) of a neuron receive input signals at the basal region, and at the apical region the neuron’s axon provides output signals.[9]
The axon initial segment is unmyelinated and contains a specialized complex of proteins. It is between approximately 20 and 60 µm in length and functions as the site of action potential initiation.[10][11] Both the position on the axon and the length of the AIS can change showing a degree of plasticity that can fine-tune the neuronal output.[10][12] A longer AIS is associated with a greater excitability.[12] Plasticity is also seen in the ability of the AIS to change its distribution and to maintain the activity of neural circuitry at a constant level.[13]
The AIS is highly specialized for the fast conduction of nerve impulses. This is achieved by a high concentration of voltage-gated sodium channels in the initial segment where the action potential is initiated.[13] The ion channels are accompanied by a high number of cell adhesion molecules and scaffolding proteins that anchor them to the cytoskeleton.[10] Interactions with ankyrin G are important as it is the major organizer in the AIS.[10]
Axonal transport[edit]
The axoplasm is the equivalent of cytoplasm in the cell. Microtubules form in the axoplasm at the axon hillock. They are arranged along the length of the axon, in overlapping sections, and all point in the same direction – towards the axon terminals.[14] This is noted by the positive endings of the microtubules. This overlapping arrangement provides the routes for the transport of different materials from the cell body.[14] Studies on the axoplasm has shown the movement of numerous vesicles of all sizes to be seen along cytoskeletal filaments – the microtubules, and neurofilaments, in both directions between the axon and its terminals and the cell body.
Outgoing anterograde transport from the cell body along the axon, carries mitochondria and membrane proteins needed for growth to the axon terminal. Ingoing retrograde transport carries cell waste materials from the axon terminal to the cell body.[15] Outgoing and ingoing tracks use different sets of motor proteins.[14] Outgoing transport is provided by kinesin, and ingoing return traffic is provided by dynein. Dynein is minus-end directed.[15] There are many forms of kinesin and dynein motor proteins, and each is thought to carry a different cargo.[14] The studies on transport in the axon led to the naming of kinesin.[14]
Myelination[edit]

TEM of a myelinated axon in cross-section.

In the nervous system, axons may be myelinated, or unmyelinated. This is the provision of an insulating layer, called a myelin sheath. The myelin membrane is unique in its relatively high lipid to protein ratio.[16]
In the peripheral nervous system axons are myelinated by glial cells known as Schwann cells. In the central nervous system the myelin sheath is provided by another type of glial cell, the oligodendrocyte. Schwann cells myelinate a single axon. An oligodendrocyte can myelinate up to 50 axons.[17]
The composition of myelin is different in the two types. In the CNS the major myelin protein is proteolipid protein, and in the PNS it is myelin basic protein.
Nodes of Ranvier[edit]
Nodes of Ranvier (also known as myelin sheath gaps) are short unmyelinated segments of a myelinated axon, which are found periodically interspersed between segments of the myelin sheath. Therefore, at the point of the node of Ranvier, the axon is reduced in diameter.[18] These nodes are areas where action potentials can be generated. In saltatory conduction, electrical currents produced at each node of Ranvier are conducted with little attenuation to the next node in line, where they remain strong enough to generate another action potential. Thus in a myelinated axon, action potentials effectively «jump» from node to node, bypassing the myelinated stretches in between, resulting in a propagation speed much faster than even the fastest unmyelinated axon can sustain.
Axon terminals[edit]
An axon can divide into many branches called telodendria (Greek for ‘end of tree’). At the end of each telodendron is an axon terminal (also called a synaptic bouton, or terminal bouton). Axon terminals contain synaptic vesicles that store the neurotransmitter for release at the synapse. This makes multiple synaptic connections with other neurons possible. Sometimes the axon of a neuron may synapse onto dendrites of the same neuron, when it is known as an autapse.
Action potentials[edit]
| Structure of a typical chemical synapse |
|---|
|
Postsynaptic Voltage- Synaptic Neurotransmitter Receptor Neurotransmitter Axon terminal Synaptic cleft Dendrite |
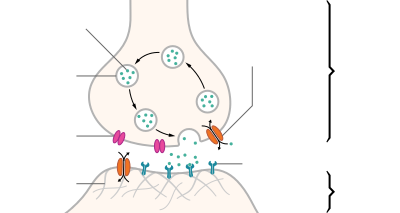
Most axons carry signals in the form of action potentials, which are discrete electrochemical impulses that travel rapidly along an axon, starting at the cell body and terminating at points where the axon makes synaptic contact with target cells. The defining characteristic of an action potential is that it is «all-or-nothing» – every action potential that an axon generates has essentially the same size and shape. This all-or-nothing characteristic allows action potentials to be transmitted from one end of a long axon to the other without any reduction in size. There are, however, some types of neurons with short axons that carry graded electrochemical signals, of variable amplitude.
When an action potential reaches a presynaptic terminal, it activates the synaptic transmission process. The first step is rapid opening of calcium ion channels in the membrane of the axon, allowing calcium ions to flow inward across the membrane. The resulting increase in intracellular calcium concentration causes synaptic vesicles (tiny containers enclosed by a lipid membrane) filled with a neurotransmitter chemical to fuse with the axon’s membrane and empty their contents into the extracellular space. The neurotransmitter is released from the presynaptic nerve through exocytosis. The neurotransmitter chemical then diffuses across to receptors located on the membrane of the target cell. The neurotransmitter binds to these receptors and activates them. Depending on the type of receptors that are activated, the effect on the target cell can be to excite the target cell, inhibit it, or alter its metabolism in some way. This entire sequence of events often takes place in less than a thousandth of a second. Afterward, inside the presynaptic terminal, a new set of vesicles is moved into position next to the membrane, ready to be released when the next action potential arrives. The action potential is the final electrical step in the integration of synaptic messages at the scale of the neuron.[5]

(A) pyramidal cell, interneuron, and short durationwaveform (Axon), overlay of the three average waveforms;
(B) Average and standard error of peak-trough time for pyramidal cells interneurons, and putative axons;
(C) Scatter plot of signal to noise ratios for individual units againstpeak-trough time for axons, pyramidal cells (PYR) and interneurons (INT).
Extracellular recordings of action potential propagation in axons has been demonstrated in freely moving animals. While extracellular somatic action potentials have been used to study cellular activity in freely moving animals such as place cells, axonal activity in both white and gray matter can also be recorded. Extracellular recordings of axon action potential propagation is distinct from somatic action potentials in three ways: 1. The signal has a shorter peak-trough duration (~150μs) than of pyramidal cells (~500μs) or interneurons (~250μs). 2. The voltage change is triphasic. 3. Activity recorded on a tetrode is seen on only one of the four recording wires. In recordings from freely moving rats, axonal signals have been isolated in white matter tracts including the alveus and the corpus callosum as well hippocampal gray matter.[19]
In fact, the generation of action potentials in vivo is sequential in nature, and these sequential spikes constitute the digital codes in the neurons. Although previous studies indicate an axonal origin of a single spike evoked by short-term pulses, physiological signals in vivo trigger the initiation of sequential spikes at the cell bodies of the neurons.[20][21]
In addition to propagating action potentials to axonal terminals, the axon is able to amplify the action potentials, which makes sure a secure propagation of sequential action potentials toward the axonal terminal. In terms of molecular mechanisms, voltage-gated sodium channels in the axons possess lower threshold and shorter refractory period in response to short-term pulses.[22]
Development and growth[edit]
Development[edit]
The development of the axon to its target, is one of the six major stages in the overall development of the nervous system.[23] Studies done on cultured hippocampal neurons suggest that neurons initially produce multiple neurites that are equivalent, yet only one of these neurites is destined to become the axon.[24] It is unclear whether axon specification precedes axon elongation or vice versa,[25] although recent evidence points to the latter. If an axon that is not fully developed is cut, the polarity can change and other neurites can potentially become the axon. This alteration of polarity only occurs when the axon is cut at least 10 μm shorter than the other neurites. After the incision is made, the longest neurite will become the future axon and all the other neurites, including the original axon, will turn into dendrites.[26] Imposing an external force on a neurite, causing it to elongate, will make it become an axon.[27] Nonetheless, axonal development is achieved through a complex interplay between extracellular signaling, intracellular signaling and cytoskeletal dynamics.
[edit]
The extracellular signals that propagate through the extracellular matrix surrounding neurons play a prominent role in axonal development.[28] These signaling molecules include proteins, neurotrophic factors, and extracellular matrix and adhesion molecules.
Netrin (also known as UNC-6) a secreted protein, functions in axon formation. When the UNC-5 netrin receptor is mutated, several neurites are irregularly projected out of neurons and finally a single axon is extended anteriorly.[29][30][31][32] The neurotrophic factors – nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NTF3) are also involved in axon development and bind to Trk receptors.[33]
The ganglioside-converting enzyme plasma membrane ganglioside sialidase (PMGS), which is involved in the activation of TrkA at the tip of neutrites, is required for the elongation of axons. PMGS asymmetrically distributes to the tip of the neurite that is destined to become the future axon.[34]
Intracellular signaling[edit]
During axonal development, the activity of PI3K is increased at the tip of destined axon. Disrupting the activity of PI3K inhibits axonal development. Activation of PI3K results in the production of phosphatidylinositol (3,4,5)-trisphosphate (PtdIns) which can cause significant elongation of a neurite, converting it into an axon. As such, the overexpression of phosphatases that dephosphorylate PtdIns leads into the failure of polarization.[28]
Cytoskeletal dynamics[edit]
The neurite with the lowest actin filament content will become the axon. PGMS concentration and f-actin content are inversely correlated; when PGMS becomes enriched at the tip of a neurite, its f-actin content is substantially decreased.[34] In addition, exposure to actin-depolimerizing drugs and toxin B (which inactivates Rho-signaling) causes the formation of multiple axons. Consequently, the interruption of the actin network in a growth cone will promote its neurite to become the axon.[35]
Growth[edit]

Axon of nine-day-old mouse with growth cone visible
Growing axons move through their environment via the growth cone, which is at the tip of the axon. The growth cone has a broad sheet-like extension called a lamellipodium which contain protrusions called filopodia. The filopodia are the mechanism by which the entire process adheres to surfaces and explores the surrounding environment. Actin plays a major role in the mobility of this system. Environments with high levels of cell adhesion molecules (CAMs) create an ideal environment for axonal growth. This seems to provide a «sticky» surface for axons to grow along. Examples of CAMs specific to neural systems include N-CAM, TAG-1 – an axonal glycoprotein[36] – and MAG, all of which are part of the immunoglobulin superfamily. Another set of molecules called extracellular matrix-adhesion molecules also provide a sticky substrate for axons to grow along. Examples of these molecules include laminin, fibronectin, tenascin, and perlecan. Some of these are surface bound to cells and thus act as short range attractants or repellents. Others are difusible ligands and thus can have long range effects.
Cells called guidepost cells assist in the guidance of neuronal axon growth. These cells that help axon guidance, are typically other neurons that are sometimes immature. When the axon has completed its growth at its connection to the target, the diameter of the axon can increase by up to five times, depending on the speed of conduction required.[37]
It has also been discovered through research that if the axons of a neuron were damaged, as long as the soma (the cell body of a neuron) is not damaged, the axons would regenerate and remake the synaptic connections with neurons with the help of guidepost cells. This is also referred to as neuroregeneration.[38]
Nogo-A is a type of neurite outgrowth inhibitory component that is present in the central nervous system myelin membranes (found in an axon). It has a crucial role in restricting axonal regeneration in adult mammalian central nervous system. In recent studies, if Nogo-A is blocked and neutralized, it is possible to induce long-distance axonal regeneration which leads to enhancement of functional recovery in rats and mouse spinal cord. This has yet to be done on humans.[39] A recent study has also found that macrophages activated through a specific inflammatory pathway activated by the Dectin-1 receptor are capable of promoting axon recovery, also however causing neurotoxicity in the neuron.[40]
Length regulation[edit]
Axons vary largely in length from a few micrometers up to meters in some animals. This emphasizes that there must be a cellular length regulation mechanism allowing the neurons both to sense the length of their axons and to control their growth accordingly. It was discovered that motor proteins play an important role in regulating the length of axons.[41] Based on this observation, researchers developed an explicit model for axonal growth describing how motor proteins could affect the axon length on the molecular level.[42][43][44][45] These studies suggest that motor proteins carry signaling molecules from the soma to the growth cone and vice versa whose concentration oscillates in time with a length-dependent frequency.
Classification[edit]
The axons of neurons in the human peripheral nervous system can be classified based on their physical features and signal conduction properties. Axons were known to have different thicknesses (from 0.1 to 20 µm)[3] and these differences were thought to relate to the speed at which an action potential could travel along the axon – its conductance velocity. Erlanger and Gasser proved this hypothesis, and identified several types of nerve fiber, establishing a relationship between the diameter of an axon and its nerve conduction velocity. They published their findings in 1941 giving the first classification of axons.
Axons are classified in two systems. The first one introduced by Erlanger and Gasser, grouped the fibers into three main groups using the letters A, B, and C. These groups, group A, group B, and group C include both the sensory fibers (afferents) and the motor fibers (efferents). The first group A, was subdivided into alpha, beta, gamma, and delta fibers – Aα, Aβ, Aγ, and Aδ. The motor neurons of the different motor fibers, were the lower motor neurons – alpha motor neuron, beta motor neuron, and gamma motor neuron having the Aα, Aβ, and Aγ nerve fibers, respectively.
Later findings by other researchers identified two groups of Aa fibers that were sensory fibers. These were then introduced into a system that only included sensory fibers (though some of these were mixed nerves and were also motor fibers). This system refers to the sensory groups as Types and uses Roman numerals: Type Ia, Type Ib, Type II, Type III, and Type IV.
Motor[edit]
Lower motor neurons have two kind of fibers:
| Type | Erlanger-Gasser Classification |
Diameter (µm) |
Myelin | Conduction velocity (meters/second) |
Associated muscle fibers |
|---|---|---|---|---|---|
| Alpha (α) motor neuron | Aα | 13–20 | Yes | 80–120 | Extrafusal muscle fibers |
| Beta (β) motor neuron | Aβ | ||||
| Gamma (γ) motor neuron | Aγ | 5-8 | Yes | 4–24[46][47] | Intrafusal muscle fibers |
Sensory[edit]
Different sensory receptors innervate different types of nerve fibers. Proprioceptors are innervated by type Ia, Ib and II sensory fibers, mechanoreceptors by type II and III sensory fibers and nociceptors and thermoreceptors by type III and IV sensory fibers.
| Type | Erlanger-Gasser Classification |
Diameter (µm) |
Myelin | Conduction velocity (m/s) |
Associated sensory receptors | Proprioceptors | Mechanoceptors | Nociceptors and thermoreceptors |
|---|---|---|---|---|---|---|---|---|
| Ia | Aα | 13–20 | Yes | 80–120 | Primary receptors of muscle spindle (annulospiral ending) | ✔ | ||
| Ib | Aα | 13–20 | Yes | 80–120 | Golgi tendon organ | |||
| II | Aβ | 6–12 | Yes | 33–75 | Secondary receptors of muscle spindle (flower-spray ending). All cutaneous mechanoreceptors |
✔ | ||
| III | Aδ | 1–5 | Thin | 3–30 | Free nerve endings of touch and pressure Nociceptors of lateral spinothalamic tract Cold thermoreceptors |
✔ | ||
| IV | C | 0.2–1.5 | No | 0.5–2.0 | Nociceptors of anterior spinothalamic tract Warmth receptors |
Autonomic[edit]
The autonomic nervous system has two kinds of peripheral fibers:
| Type | Erlanger-Gasser Classification |
Diameter (µm) |
Myelin[48] | Conduction velocity (m/s) |
|---|---|---|---|---|
| preganglionic fibers | B | 1–5 | Yes | 3–15 |
| postganglionic fibers | C | 0.2–1.5 | No | 0.5–2.0 |
Clinical significance[edit]
In order of degree of severity, injury to a nerve can be described as neurapraxia, axonotmesis, or neurotmesis.
Concussion is considered a mild form of diffuse axonal injury.[49] Axonal injury can also cause central chromatolysis. The dysfunction of axons in the nervous system is one of the major causes of many inherited neurological disorders that affect both peripheral and central neurons.[5]
When an axon is crushed, an active process of axonal degeneration takes place at the part of the axon furthest from the cell body. This degeneration takes place quickly following the injury, with the part of the axon being sealed off at the membranes and broken down by macrophages. This is known as Wallerian degeneration.[50] Dying back of an axon can also take place in many neurodegenerative diseases, particularly when axonal transport is impaired, this is known as Wallerian-like degeneration.[51] Studies suggest that the degeneration happens as
a result of the axonal protein NMNAT2, being prevented from reaching all of the axon.[52]
Demyelination of axons causes the multitude of neurological symptoms found in the disease multiple sclerosis.
Dysmyelination is the abnormal formation of the myelin sheath. This is implicated in several leukodystrophies, and also in schizophrenia.[53][54][55]
A severe traumatic brain injury can result in widespread lesions to nerve tracts damaging the axons in a condition known as diffuse axonal injury. This can lead to a persistent vegetative state.[56] It has been shown in studies on the rat that axonal damage from a single mild traumatic brain injury, can leave a susceptibility to further damage, after repeated mild traumatic brain injuries.[57]
A nerve guidance conduit is an artificial means of guiding axon growth to enable neuroregeneration, and is one of the many treatments used for different kinds of nerve injury.
History[edit]
German anatomist Otto Friedrich Karl Deiters is generally credited with the discovery of the axon by distinguishing it from the dendrites.[5] Swiss Rüdolf Albert von Kölliker and German Robert Remak were the first to identify and characterize the axon initial segment. Kölliker named the axon in 1896.[58] Louis-Antoine Ranvier was the first to describe the gaps or nodes found on axons and for this contribution these axonal features are now commonly referred to as the nodes of Ranvier. Santiago Ramón y Cajal, a Spanish anatomist, proposed that axons were the output components of neurons, describing their functionality.[5] Joseph Erlanger and Herbert Gasser earlier developed the classification system for peripheral nerve fibers,[59] based on axonal conduction velocity, myelination, fiber size etc. Alan Hodgkin and Andrew Huxley also employed the squid giant axon (1939) and by 1952 they had obtained a full quantitative description of the ionic basis of the action potential, leading to the formulation of the Hodgkin–Huxley model. Hodgkin and Huxley were awarded jointly the Nobel Prize for this work in 1963. The formulae detailing axonal conductance were extended to vertebrates in the Frankenhaeuser–Huxley equations. The understanding of the biochemical basis for action potential propagation has advanced further, and includes many details about individual ion channels.
Other animals[edit]
The axons in invertebrates have been extensively studied. The longfin inshore squid, often used as a model organism has the longest known axon.[60] The giant squid has the largest axon known. Its size ranges from 0.5 (typically) to 1 mm in diameter and is used in the control of its jet propulsion system. The fastest recorded conduction speed of 210 m/s, is found in the ensheathed axons of some pelagic Penaeid shrimps[61] and the usual range is between 90 and 200 meters/s[62] (cf 100–120 m/s for the fastest myelinated vertebrate axon.)
In other cases as seen in rat studies an axon originates from a dendrite; such axons are said to have «dendritic origin». Some axons with dendritic origin similarly have a «proximal» initial segment that starts directly at the axon origin, while others have a «distal» initial segment, discernibly separated from the axon origin.[63] In many species some of the neurons have axons that emanate from the dendrite and not from the cell body, and these are known as axon-carrying dendrites.[1] In many cases, an axon originates at an axon hillock on the soma; such axons are said to have «somatic origin». Some axons with somatic origin have a «proximal» initial segment adjacent the axon hillock, while others have a «distal» initial segment, separated from the soma by an extended axon hillock.[63]
See also[edit]
- Electrophysiology
- Ganglionic eminence
- Giant axonal neuropathy
- Neuronal tracing
- Pioneer axon
References[edit]
- ^ a b Triarhou LC (2014). «Axons emanating from dendrites: phylogenetic repercussions with Cajalian hues». Frontiers in Neuroanatomy. 8: 133. doi:10.3389/fnana.2014.00133. PMC 4235383. PMID 25477788.
- ^ Yau KW (December 1976). «Receptive fields, geometry and conduction block of sensory neurones in the central nervous system of the leech». The Journal of Physiology. 263 (3): 513–38. doi:10.1113/jphysiol.1976.sp011643. PMC 1307715. PMID 1018277.
- ^ a b c Squire, Larry (2013). Fundamental neuroscience (4th ed.). Amsterdam: Elsevier/Academic Press. pp. 61–65. ISBN 978-0-12-385-870-2.
- ^ a b c Luders E, Thompson PM, Toga AW (August 2010). «The development of the corpus callosum in the healthy human brain». The Journal of Neuroscience. 30 (33): 10985–90. doi:10.1523/JNEUROSCI.5122-09.2010. PMC 3197828. PMID 20720105.
- ^ a b c d e Debanne D, Campanac E, Bialowas A, Carlier E, Alcaraz G (April 2011). «Axon physiology» (PDF). Physiological Reviews. 91 (2): 555–602. doi:10.1152/physrev.00048.2009. PMID 21527732. S2CID 13916255.
- ^ Nelson AD, Jenkins PM (2017). «Axonal Membranes and Their Domains: Assembly and Function of the Axon Initial Segment and Node of Ranvier». Frontiers in Cellular Neuroscience. 11: 136. doi:10.3389/fncel.2017.00136. PMC 5422562. PMID 28536506.
- ^ Leterrier C, Clerc N, Rueda-Boroni F, Montersino A, Dargent B, Castets F (2017). «Ankyrin G Membrane Partners Drive the Establishment and Maintenance of the Axon Initial Segment». Frontiers in Cellular Neuroscience. 11: 6. doi:10.3389/fncel.2017.00006. PMC 5266712. PMID 28184187.
- ^ Leterrier C (February 2018). «The Axon Initial Segment: An Updated Viewpoint». The Journal of Neuroscience. 38 (9): 2135–2145. doi:10.1523/jneurosci.1922-17.2018. PMC 6596274. PMID 29378864.
- ^ Rasband MN (August 2010). «The axon initial segment and the maintenance of neuronal polarity». Nature Reviews. Neuroscience. 11 (8): 552–62. doi:10.1038/nrn2852. PMID 20631711. S2CID 23996233.
- ^ a b c d Jones SL, Svitkina TM (2016). «Axon Initial Segment Cytoskeleton: Architecture, Development, and Role in Neuron Polarity». Neural Plasticity. 2016: 6808293. doi:10.1155/2016/6808293. PMC 4967436. PMID 27493806.
- ^ Clark BD, Goldberg EM, Rudy B (December 2009). «Electrogenic tuning of the axon initial segment». The Neuroscientist. 15 (6): 651–68. doi:10.1177/1073858409341973. PMC 2951114. PMID 20007821.
- ^ a b Yamada R, Kuba H (2016). «Structural and Functional Plasticity at the Axon Initial Segment». Frontiers in Cellular Neuroscience. 10: 250. doi:10.3389/fncel.2016.00250. PMC 5078684. PMID 27826229.
- ^ a b Susuki K, Kuba H (March 2016). «Activity-dependent regulation of excitable axonal domains». The Journal of Physiological Sciences. 66 (2): 99–104. doi:10.1007/s12576-015-0413-4. PMID 26464228. S2CID 18862030.
- ^ a b c d e Alberts B (2004). Essential cell biology: an introduction to the molecular biology of the cell (2nd ed.). New York: Garland. pp. 584–587. ISBN 978-0-8153-3481-1.
- ^ a b Alberts B (2002). Molecular biology of the cell (4th ed.). New York: Garland. pp. 979–981. ISBN 978-0-8153-4072-0.
- ^ Ozgen, H; Baron, W; Hoekstra, D; Kahya, N (September 2016). «Oligodendroglial membrane dynamics in relation to myelin biogenesis». Cellular and Molecular Life Sciences. 73 (17): 3291–310. doi:10.1007/s00018-016-2228-8. PMC 4967101. PMID 27141942.
- ^ Sadler, T. (2010). Langman’s medical embryology (11th ed.). Philadelphia: Lippincott William & Wilkins. p. 300. ISBN 978-0-7817-9069-7.
- ^ Hess A, Young JZ (November 1952). «The nodes of Ranvier». Proceedings of the Royal Society of London. Series B, Biological Sciences. Series B. 140 (900): 301–20. Bibcode:1952RSPSB.140..301H. doi:10.1098/rspb.1952.0063. JSTOR 82721. PMID 13003931. S2CID 11963512.
- ^ Robbins AA, Fox SE, Holmes GL, Scott RC, Barry JM (November 2013). «Short duration waveforms recorded extracellularly from freely moving rats are representative of axonal activity». Frontiers in Neural Circuits. 7 (181): 181. doi:10.3389/fncir.2013.00181. PMC 3831546. PMID 24348338.
- ^ Rongjing Ge, Hao Qian and Jin-Hui Wang* (2011) Molecular Brain 4(19), 1~11
- ^ Rongjing Ge, Hao Qian, Na Chen and Jin-Hui Wang* (2014) Molecular Brain 7(26):1-16
- ^ Chen N, Yu J, Qian H, Ge R, Wang JH (July 2010). «Axons amplify somatic incomplete spikes into uniform amplitudes in mouse cortical pyramidal neurons». PLOS ONE. 5 (7): e11868. Bibcode:2010PLoSO…511868C. doi:10.1371/journal.pone.0011868. PMC 2912328. PMID 20686619.
- ^ Wolpert, Lewis (2015). Principles of development (5th ed.). pp. 520–524. ISBN 978-0-19-967814-3.
- ^ Fletcher TL, Banker GA (December 1989). «The establishment of polarity by hippocampal neurons: the relationship between the stage of a cell’s development in situ and its subsequent development in culture». Developmental Biology. 136 (2): 446–54. doi:10.1016/0012-1606(89)90269-8. PMID 2583372.
- ^ Jiang H, Rao Y (May 2005). «Axon formation: fate versus growth». Nature Neuroscience. 8 (5): 544–6. doi:10.1038/nn0505-544. PMID 15856056. S2CID 27728967.
- ^ Goslin K, Banker G (April 1989). «Experimental observations on the development of polarity by hippocampal neurons in culture». The Journal of Cell Biology. 108 (4): 1507–16. doi:10.1083/jcb.108.4.1507. PMC 2115496. PMID 2925793.
- ^ Lamoureux P, Ruthel G, Buxbaum RE, Heidemann SR (November 2002). «Mechanical tension can specify axonal fate in hippocampal neurons». The Journal of Cell Biology. 159 (3): 499–508. doi:10.1083/jcb.200207174. PMC 2173080. PMID 12417580.
- ^ a b Arimura N, Kaibuchi K (March 2007). «Neuronal polarity: from extracellular signals to intracellular mechanisms». Nature Reviews. Neuroscience. 8 (3): 194–205. doi:10.1038/nrn2056. PMID 17311006. S2CID 15556921.
- ^ Neuroglia and pioneer neurons express UNC-6 to provide global and local netrin cues for guiding migrations in C. elegans
- ^ Serafini T, Kennedy TE, Galko MJ, Mirzayan C, Jessell TM, Tessier-Lavigne M (August 1994). «The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6». Cell. 78 (3): 409–24. doi:10.1016/0092-8674(94)90420-0. PMID 8062384. S2CID 22666205.
- ^ Hong K, Hinck L, Nishiyama M, Poo MM, Tessier-Lavigne M, Stein E (June 1999). «A ligand-gated association between cytoplasmic domains of UNC5 and DCC family receptors converts netrin-induced growth cone attraction to repulsion». Cell. 97 (7): 927–41. doi:10.1016/S0092-8674(00)80804-1. PMID 10399920. S2CID 18043414.
- ^ Hedgecock EM, Culotti JG, Hall DH (January 1990). «The unc-5, unc-6, and unc-40 genes guide circumferential migrations of pioneer axons and mesodermal cells on the epidermis in C. elegans». Neuron. 4 (1): 61–85. doi:10.1016/0896-6273(90)90444-K. PMID 2310575. S2CID 23974242.
- ^ Huang EJ, Reichardt LF (2003). «Trk receptors: roles in neuronal signal transduction». Annual Review of Biochemistry. 72: 609–42. doi:10.1146/annurev.biochem.72.121801.161629. PMID 12676795. S2CID 10217268.
- ^ a b Da Silva JS, Hasegawa T, Miyagi T, Dotti CG, Abad-Rodriguez J (May 2005). «Asymmetric membrane ganglioside sialidase activity specifies axonal fate». Nature Neuroscience. 8 (5): 606–15. doi:10.1038/nn1442. PMID 15834419. S2CID 25227765.
- ^ Bradke F, Dotti CG (March 1999). «The role of local actin instability in axon formation». Science. 283 (5409): 1931–4. Bibcode:1999Sci…283.1931B. doi:10.1126/science.283.5409.1931. PMID 10082468.
- ^ Furley AJ, Morton SB, Manalo D, Karagogeos D, Dodd J, Jessell TM (April 1990). «The axonal glycoprotein TAG-1 is an immunoglobulin superfamily member with neurite outgrowth-promoting activity». Cell. 61 (1): 157–70. doi:10.1016/0092-8674(90)90223-2. PMID 2317872. S2CID 28813676.
- ^ Alberts, Bruce (2015). Molecular biology of the cell (Sixth ed.). p. 947. ISBN 9780815344643.
- ^ Kunik D, Dion C, Ozaki T, Levin LA, Costantino S (2011). «Laser-based single-axon transection for high-content axon injury and regeneration studies». PLOS ONE. 6 (11): e26832. Bibcode:2011PLoSO…626832K. doi:10.1371/journal.pone.0026832. PMC 3206876. PMID 22073205.
- ^ Schwab ME (February 2004). «Nogo and axon regeneration». Current Opinion in Neurobiology. 14 (1): 118–24. doi:10.1016/j.conb.2004.01.004. PMID 15018947. S2CID 9672315.
- ^ Gensel JC, Nakamura S, Guan Z, van Rooijen N, Ankeny DP, Popovich PG (March 2009). «Macrophages promote axon regeneration with concurrent neurotoxicity». The Journal of Neuroscience. 29 (12): 3956–68. doi:10.1523/JNEUROSCI.3992-08.2009. PMC 2693768. PMID 19321792.
- ^ Myers KA, Baas PW (September 2007). «Kinesin-5 regulates the growth of the axon by acting as a brake on its microtubule array». The Journal of Cell Biology. 178 (6): 1081–91. doi:10.1083/jcb.200702074. PMC 2064629. PMID 17846176.
- ^ Rishal I, Kam N, Perry RB, Shinder V, Fisher EM, Schiavo G, Fainzilber M (June 2012). «A motor-driven mechanism for cell-length sensing». Cell Reports. 1 (6): 608–16. doi:10.1016/j.celrep.2012.05.013. PMC 3389498. PMID 22773964.
- ^ Karamched BR, Bressloff PC (May 2015). «Delayed feedback model of axonal length sensing». Biophysical Journal. 108 (9): 2408–19. Bibcode:2015BpJ…108.2408K. doi:10.1016/j.bpj.2015.03.055. PMC 4423051. PMID 25954897.
- ^ Bressloff PC, Karamched BR (2015). «A frequency-dependent decoding mechanism for axonal length sensing». Frontiers in Cellular Neuroscience. 9: 281. doi:10.3389/fncel.2015.00281. PMC 4508512. PMID 26257607.
- ^ Folz F, Wettmann L, Morigi G, Kruse K (May 2019). «Sound of an axon’s growth». Physical Review E. 99 (5–1): 050401. arXiv:1807.04799. Bibcode:2019PhRvE..99e0401F. doi:10.1103/PhysRevE.99.050401. PMID 31212501. S2CID 118682719.
- ^ Andrew BL, Part NJ (April 1972). «Properties of fast and slow motor units in hind limb and tail muscles of the rat». Quarterly Journal of Experimental Physiology and Cognate Medical Sciences. 57 (2): 213–25. doi:10.1113/expphysiol.1972.sp002151. PMID 4482075.
- ^ Russell NJ (January 1980). «Axonal conduction velocity changes following muscle tenotomy or deafferentation during development in the rat». The Journal of Physiology. 298: 347–60. doi:10.1113/jphysiol.1980.sp013085. PMC 1279120. PMID 7359413.
- ^ Pocock G, Richards CD, et al. (2004). Human Physiology (2nd ed.). New York: Oxford University Press. pp. 187–189. ISBN 978-0-19-858527-5.
- ^ Dawodu ST (16 August 2017). «Traumatic Brain Injury (TBI) — Definition, Epidemiology, Pathophysiology». Medscape. Archived from the original on 12 June 2018. Retrieved 14 July 2018.
- ^ Trauma and Wallerian Degeneration Archived 2 May 2006 at the Wayback Machine, University of California, San Francisco
- ^ Coleman MP, Freeman MR (1 June 2010). «Wallerian degeneration, wld(s), and nmnat». Annual Review of Neuroscience. 33 (1): 245–67. doi:10.1146/annurev-neuro-060909-153248. PMC 5223592. PMID 20345246.
- ^ Gilley J, Coleman MP (January 2010). «Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons». PLOS Biology. 8 (1): e1000300. doi:10.1371/journal.pbio.1000300. PMC 2811159. PMID 20126265.
- ^ Krämer-Albers EM, Gehrig-Burger K, Thiele C, Trotter J, Nave KA (November 2006). «Perturbed interactions of mutant proteolipid protein/DM20 with cholesterol and lipid rafts in oligodendroglia: implications for dysmyelination in spastic paraplegia». The Journal of Neuroscience. 26 (45): 11743–52. doi:10.1523/JNEUROSCI.3581-06.2006. PMC 6674790. PMID 17093095.
- ^ Matalon R, Michals-Matalon K, Surendran S, Tyring SK (2006). «Canavan disease: studies on the knockout mouse». N-Acetylaspartate. Adv. Exp. Med. Biol. Advances in Experimental Medicine and Biology. Vol. 576. pp. 77–93, discussion 361–3. doi:10.1007/0-387-30172-0_6. ISBN 978-0-387-30171-6. PMID 16802706. S2CID 44405442.
- ^ Tkachev D, Mimmack ML, Huffaker SJ, Ryan M, Bahn S (August 2007). «Further evidence for altered myelin biosynthesis and glutamatergic dysfunction in schizophrenia». The International Journal of Neuropsychopharmacology. 10 (4): 557–63. doi:10.1017/S1461145706007334. PMID 17291371.
- ^ «Brain Injury, Traumatic». Medcyclopaedia. GE. Archived from the original on 26 May 2011. Retrieved 20 June 2018.
- ^ Wright DK, Brady RD, Kamnaksh A, Trezise J, Sun M, McDonald SJ, et al. (October 2019). «Repeated mild traumatic brain injuries induce persistent changes in plasma protein and magnetic resonance imaging biomarkers in the rat». Scientific Reports. 9 (1): 14626. Bibcode:2019NatSR…914626W. doi:10.1038/s41598-019-51267-w. PMC 6787341. PMID 31602002.
- ^ Finger S (1994). Origins of neuroscience: a history of explorations into brain function. Oxford University Press. p. 47. ISBN 9780195146943. OCLC 27151391.
Kölliker would give the «axon» its name in 1896.
- ^ Grant G (December 2006). «The 1932 and 1944 Nobel Prizes in physiology or medicine: rewards for ground-breaking studies in neurophysiology». Journal of the History of the Neurosciences. 15 (4): 341–57. doi:10.1080/09647040600638981. PMID 16997762. S2CID 37676544.
- ^ Hellier, Jennifer L. (16 December 2014). The Brain, the Nervous System, and Their Diseases [3 volumes]. ABC-CLIO. ISBN 9781610693387. Archived from the original on 14 March 2018.
- ^ Hsu K, Terakawa S (July 1996). «Fenestration in the myelin sheath of nerve fibers of the shrimp: a novel node of excitation for saltatory conduction». Journal of Neurobiology. 30 (3): 397–409. doi:10.1002/(SICI)1097-4695(199607)30:3<397::AID-NEU8>3.0.CO;2-#. PMID 8807532.
- ^ Salzer JL, Zalc B (October 2016). «Myelination». Current Biology. 26 (20): R971–R975. doi:10.1016/j.cub.2016.07.074. PMID 27780071.
- ^ a b Höfflin F, Jack A, Riedel C, Mack-Bucher J, Roos J, Corcelli C, et al. (2017). «Heterogeneity of the Axon Initial Segment in Interneurons and Pyramidal Cells of Rodent Visual Cortex». Frontiers in Cellular Neuroscience. 11: 332. doi:10.3389/fncel.2017.00332. PMC 5684645. PMID 29170630.
External links[edit]
- Histology image: 3_09 at the University of Oklahoma Health Sciences Center – «Slide 3 Spinal cord»
Периодически, приблизительно в 15-20% случаев, по данным «нейронного теста» (аутоантитела к аутоантигенам аксонов и миелиновой оболочки — общий белок миелина) мы выявляем у больных с психическими расстройствами и, в частности, страдающих зависимостью от психоактивных веществ, патологию аксонов, которую можно рассматривать либо как следствие воспалительного процесса, либо демиелинизирующих процессов или аксонопатию метаболического характера.
Патология аксонов, вероятно, может включать в себя: их утрату, пересечение, нарушенный аксональный транспорт, повреждение оболочки, дистального или проксимального отделов этих отростков нейронов. «Незащищенные аксоны» вследствие повреждения или утраты миелиновой оболочки становятся более уязвимыми и восприимчивыми к их дальнейшему повреждению.
В психиатрии мы чаще всего встречаемся с хроническим поражением аксонов, в неврологии, например, при рассеянном склерозе — с острым. Маркерами острого повреждения аксонов считаются: дефосфолирование нейрофиламентов, нарушение аксонального транспорта, экспрессия специфических кальциевых каналов и пересечение аксонов.
Нарушение аксонального транспорта («поперечное повреждение аксонов») можно обнаружить при помощи белков, транспортирующихся по аксонам. Белки аккумулируются в поперечных срезах или в зонах нарушенного транспорта. Одним из таких белков является амилоидный белок предшественник, который синтезируется в нейронах и транспортируется на периферию вдоль аксонального цитоскелета. Некоторые исследователи полагают, что повреждение аксонов происходит при атаке на миелиновую оболочку, что аксонотоксичные компоненты присутствуют среди медиаторов воспаления, повреждающих миелиновый — олигодендроцитарный комплекс. Возможно, аксоны утрачивают трофическую поддержку, получаемую от олигодендроцитов. Скорее всего, основными мишенями аутоиммунного ответа при некоторых психических расстройствах являются олигодендроциты и миелиновые оболочки.
Олигодендроциты более восприимчивы к повреждению вследствие влияния большого числа иммунологических или токсических эффекторов, включая цитокины (TNF-альфа), реактивные виды кислорода или азота, медиаторов возбуждения (глутамат), компонентов комплимента, протеолитических и липолитических энзимов и др. агентов. Целостность аксонов в первую очередь зависит от взаимодействия аксонов и олигодендроцитов, которые являются миелинобразующими клетками и доставляющими к аксону «трофические сигналы выживания». Заключительная фаза разрушения цитоскелета аксонов заключается в притоке в него ионов Ca2+, активирующих широкий спектр протеиназ, например, кальпаина, что и приводит к деинтеграции аксона (этот процесс можно предотвратить с помощью блокады входа в аксон Na+ или Ca+).