 Диффузное аксональное повреждение головного мозга (далее ДАП) – это черепно-мозговая травма, результатом которой является разрыв или повреждение аксонов (отростков нервных клеток), сообщающих нервные импульсы клеткам ЦНС, органам и тканям.
Диффузное аксональное повреждение головного мозга (далее ДАП) – это черепно-мозговая травма, результатом которой является разрыв или повреждение аксонов (отростков нервных клеток), сообщающих нервные импульсы клеткам ЦНС, органам и тканям.
ДАП, зачастую, ведет к коме, в результате которой человек может перейти в вегетативное состояние.
Диффузия головного мозга чаще происходит с молодыми людьми, которые попадают в дорожно-транспортные происшествия, становятся жертвами драк и избиений с повреждением головного мозга, а также детьми, у которых и кома намного глубже, и неврологические нарушения грубее.
Вариант этой ЧМТ был впервые описан в 1956 за рубежом, а само название появилось в 1982 году. Состояние при ДАП тяжелое, протекающее в длительной коме, которая сразу возникает после травмы и характеризуется длительным течением.
Аксоны, поврежденные или разорванные в результате ЧМТ, и мелкие кровоизлияния равномерно распределяются по церебральным структурам, при этом нарушается иннервация всех зависимых органов. Самые распространенные места повреждений:
- ствол мозга;
- белое вещество;
- мозольное тело;
- волокна перивентрикулярные.
Диффузная травма головного мозга – это всегда серьезнейшее состояние с перспективой перехода в вегетативное состояние, нередок и смертельный исход.
Причины и морфология ДАП
Самые распространенные причины ЧМТ при ДАП:
- сильный ушиб о лобовые стекла в ДТП;
- падение;
- удар тяжелым предметом;
- синдром сотрясения у детей, при котором мозг подвергается сильному ушибу впоследствии резкой тряски, а также избиения, падения.
Синдром ДАП является результатом ушибов, вызванных угловым ускорением головы. При этом может и не быть прямого столкновения с объектом травмы.
Вследствие этого у некоторых пациентов не наблюдается переломов черепа и других видимых ранений, что несколько затрудняет диагностику. Статистика говорит о том, что именно повреждение по косой ведет к повреждению аксонов и возникновению ДАП.
Морфологически для этой травмы характерны три следующих очага повреждения:
- головной мозг;
- мозольное тело;
- стволовой мозг и диффузно распределенные разрывы.
Первые два очаговых признака – макроскопические, которые располагаются в виде гематомы до пяти мм, выглядящие как надорванная ткань с окровавленными краями. Через несколько дней после ЧМТ, очаг пигментируется и затем рубцуется. Рана в мозолистом теле может разрешиться образованием сосудистой кисты.

Аксональные нарушения при травме
Особенности клинической картины
Кома при диффузном повреждении головного мозга продолжается до трех недель и проявляется следующими симптомами:
- рефлекс зрачка нарушается;

- происходит паралич взгляда;
- дыхательный ритм меняется;
- повышается тонус мышц;
- появляется парез ног и рук;
- диагностируется гипертония;
- появляется фебрильная или субфебрильная температура и другие вегетативные расстройства.

Когда пациент с ДАП выходит из комы, он оказывается в вегетативном состоянии со следующими симптомами:
- глаза открываются на раздражители или самостоятельно;
- взгляд не сконцентрирован и не следует за движущимися предметами.
Вегетативное состояние, сопровождающееся нарушением рефлексов и симптомами разъединения функций мозговых полушарий, может продолжаться, в среднем, от нескольких дней до нескольких лет. Чем больше нахождение в нем, тем скорее появляются такие признаки полиневропатии, как:
- ослабление кистей рук;
- хаотическое движение мышц;
- расстройства нейротрофики;
- учащение пульса;
- отеки;
- тахипноэ и др.
После выхода из вегетативного состояния личность выпадает. Основные признаки нарушений после выхода:
- экстрапирамидные расстройства;
- расстройства психики, проявляющиеся отсутствием интереса к окружающей пациента действительности, агрессия, амнезия, слабоумие.
Степени поражения
Диффузное аксональное повреждение головного мозга бывает трех степеней тяжести:
- легкой – продолжительность комы от 6 до 24 часов, ЧМТ незначительна;
- средней– кома больше 24 часов, но черепно-мозговая травма умеренная;
- тяжелой – длительная кома, поражения значительные, происходит сдавливание мозга.
Тяжелая степень характеризуется массовым повреждением аксонов, которое приводят к кровоизлияниям в мозг. ДАП при этом может приводить к коме, продолжающейся годами и летальному исходу.
Начальное состояние мозга после выхода из такой комы восстановить не представляется возможным, единицы вернулись к более-менее нормальной жизни после таких повреждений.
Постановка диагноза
Диагноз диффузное поражение головного мозга ставится после компьютерной томографии, результаты которой в остром периоде отличаются увеличением объема мозговых полушарий, уменьшением или сдавливанием боковых расстояний и основания мозга. В белом веществе, стволе и мозолистом теле находятся мелкие кровоизлияния.
При обследовании наблюдается молниеносное развитие признаков ДАП и дегенерации.
ЭЭГ при синдроме ДАП выявляет изменения в подкорке и стволе мозга, диэнцефальный синдром. В анализе крови отмечается резкое повышение серотонина, значительное уменьшение дофамина и скачок адреналина, что предполагает терапию, направленную на снижение симпатико-адреналиновых симптомов.
В результате КТ определяют повышено внутричерепное давление или, наоборот, понижено либо отсутствует. В таком случае подключают датчик. Если на КТ отток ликвора в норме, то и внутричерепное давление будет в норме.

Поддержка состояния пострадавшего
После диффузной травмы головного мозга нередко диагностируются субдуральные ликворные скопления над большими полушариями мозга, которые в дальнейшем рассасываются, удалять хирургически их не надо.
Аксональное повреждение головного мозга чаще всего лечится консервативно. Нейрохирургическая операция проводится при сочетании разрывов и повреждений аксонов с очаговыми повреждениями, усиливающими сдавливание и провоцирующими гидроцефальный синдром.
В коме пациента подключают к ИВЛ, кормят парентерально и вводят следующие лекарства:
- для установления правильного кислотно-щелочного и водно-электролитного баланса;
- ноотропные и вазоактивные;
- устраняющие гипертензию или гипотонию;
- антибиотики для исключения сопутствующих инфекций.
Для возобновления психэмоциональной сферы вводят прием психостимуляторов.
После выхода из комы:
- вводят ноотропы и сосудистые лекарства для нормализации и улучшения состояния ЦНС, ноотропы также важны для последующей реабилитации;
- назначают препараты для улучшения метаболизма и биостимуляторы;
- проводят лечебную физкультуру для профилактики парезов;
- пациент занимается с логопедом.
Гормональные препараты при ДАП не назначают по ненадобности. После операции, если она все же состоялась (произошло сдавление головного мозга при сопутствующих травмах), вводят препараты, препятствующие образованию отеков, сосудистые средства, ноотропы, антихолинэстеразные, психотропные (во избежание развития агрессии и депрессии) и нейромедиаторы.
В период восстановления проводится такая же терапия, как и после выхода из комы.
Исход тяжелой травмы и ее последствия
Прогноз и последствия диффузного аксонального повреждения зависит от степени поражения аксонов головного мозга и тяжести вторичных признаков, таких как повышенное внутричерепное давление, гипергидроз, разбухание мозговых оболочек, психические нарушения, развитие слабоумия и т. д.
Исход также зависит от того как помогают лечебные методы, направленные на ликвидацию последствий ДАП – вторичные повреждения и осложнения.
Прогноз предполагает, что чем больше человек находился в коматозном состоянии, тем более риск развития неблагоприятных поражений, вплоть до летального исхода. Шансов на восстановление будет также минимум.
 Но, нужно сказать, что иногда восстановиться полностью или почти полностью, вернуть психические функции, вернуться к нормальной деятельности, убрать все неврологические нарушения можно, даже если человек находился в коме третьей степени (тяжелой), а после продолжительное время пребывал в вегетативном состоянии. Тенденция к самовосстановлению всегда присутствует у мозга, известны и более тяжелые нарушения, при которых он восстанавливался.
Но, нужно сказать, что иногда восстановиться полностью или почти полностью, вернуть психические функции, вернуться к нормальной деятельности, убрать все неврологические нарушения можно, даже если человек находился в коме третьей степени (тяжелой), а после продолжительное время пребывал в вегетативном состоянии. Тенденция к самовосстановлению всегда присутствует у мозга, известны и более тяжелые нарушения, при которых он восстанавливался.
Но, к сожалению, чаще у выживших людей последующее течение синдрома ДАП может идти по двум сценариям:
- выход из коматозного состояния;
- переход в вегетативное состояние.
При первом варианте глаза больного открываются, и происходит слежение за предметами и фиксация взгляда на объекте. Это может иметь как спонтанный выход, так и направляемый организованными раздражителями, звуком и болевыми манипуляциями.
Затем пациент восстанавливает сознание, выполняет обращенные к нему просьбы, словесный багаж расширяется, он начинает общаться. Неврологические патологии при этом медленно регрессируют.
У больных, которые вышли из вегетативного состояния, развиваются экстрапирамидальные симптомы, сопровождающиеся психическими нарушениями (слабоумие, лабильность настроения, аспонтанность, спутанность сознания). При втором варианте летальный исход через определенное время неизбежен из-за истощения нейромедиаторов и соматических осложнений.
Современные исследования подтверждают регенерацию аксонов у детей и молодых людей, у которых мозг еще не завершил формирование. Происходит восстановление неврологических и психических процессов. При продолжительной коме оно проблематично, инвалидизация гарантирована.
- Авторы
- Резюме
- Файлы
- Ключевые слова
- Литература
Ефимов А.А.
1
Савенкова Е.Н.
1
Алексеев Ю.Д.
1
Райкова К.А.
1
Коротина О.С.
1
Корсак В.О.
1
1 ФГБОУ ВО «Саратовский государственный медицинский университет им. В.И. Разумовского Минздрава России»
Диффузное аксональное повреждение (ДАП) головного мозга остается предметом исследования с позиций разных специальностей и не теряет своей актуальности, как вариант тяжелой формы черепно-мозговой травмы. Проблема судебно-медицинской диагностики ДАП в настоящее время требует обобщения и анализа теоретических данных для их практического применения и выработки алгоритма постановки диагноза этой сложной патологии. Несоответствие тяжести состояния пострадавшего и морфологических проявлений при отсутствии стандартизированных критериев судебно-медицинской верификации диктует необходимость дальнейшего изучения данного вопроса. В обзоре представлены современные сведения о механизме, патоморфологической и клинической картине, а также существующих методах судебно-медицинской диагностики ДАП. Проанализирована возможность использования различных веществ в качестве индикаторов повреждения головного мозга (таких как β-APP белки, белок S-100, нейрон-специфическая енолаза, глиальный фибриллярный кислый протеин, альфа-II-спектрин, расщепленный тау-белок). Представленные данные научных работ свидетельствуют о необходимости комплексного применения различных методов исследования (анализ клинической картины, морфологических, гистологических изменений и биомаркеров) в случаях с подозрением на ДАП. Это позволит повысить обоснованность судебно-медицинской диагностики данной патологии в случаях тяжелых черепно-мозговых травм.
диффузное аксональное повреждение
черепно-мозговая травма
судебно-медицинская экспертная оценка дап
1. Арефьева Е.Г., Гатин Д.В., Мошнегуц С.В., Рохленко О.В., Ушаков А.В., Шалякин К.Л. Диффузное аксональное повреждение: кт-картина и клинические наблюдения// Медицинская визуализация. 2012. № 1. С. 45-50.
2. Арушанян М.Ю. Диффузное аксональное повреждение головного мозга // Евразийское Научное Объединение. 2019. № 6-3 (52). С. 173-176.
3. Лихтерман Л.Б. Диффузное аксональное повреждение головного мозга // Неврология и ревматология. Приложение к журналу ConsiliumMedicum. 2016. № 1. С. 44-51.
4. Храпов Ю.В., Поройский С.В. Роль биомаркеров повреждения вещества головного мозга в диагностике, оценке эффективности лечения и прогнозировании исходов тяжелой черепно-мозговой травмы. // Волгоградский научно-медицинский журнал. 2013. № 3(39). C. 10–20.
5. SenuakovaO.V., GalanineV.E., KrylovA.S., PetraikinA.V., AkhadovT.A., SidorinS.V. Diffuseaxonalinjurylesionsegmentationusingcontouringalgorithm. In: ProceedingsofGraphiCon. 2011. P 84–87.
6. Vieira R.М., Paiva, W.S., Oliveira D. V., Teixeira M. J., Andrade A. F. Diffuse Axonal Injury: Epidemiology.Outcome and Associated Risk Factors. Frontiers in Neurology. 2016. vol. 7.no. 178. Р. 1-12.
7. Xie Y., Tao X. White matter lesion segmentation using machine learning and weakly labeled MR images. In: Proceeding of SPIE. 2011.no. 7962. P. 132-141.
8. Петряйкин А.В., Ахадов Т.А., Сенюкова О.В., Крылов А.С. Алгоритм идентификации очагов диффузно-аксонального повреждения у больных с черепно-мозговой травмой //Нейроимиджинг и магнитоэнцефалография: фундаментальные исследования и клиническая практика: сборник трудов международного симпозиума. М., 2012. С 92–94.
9. Сафронова Е.С. Диффузное аксональное повреждение мозга — современные представления о патогенетических механизмах и перспективах фармакотерапии // Забайкальский медицинский вестник. 2013. № 1. С. 206-213.
10. Сафронова Е.С., Юнцев С.В., Белозерцев Ю.А. Поиск противосудорожных средств с многофакторным нейропротекторным действием для терапии диффузной аксональной травмы мозга // Вестник Забайкальского государственного университета. 2013. № 4. С. 59-65.
11. Сафронова Е.С., Белозерцев Ю.А., Юнцов С.В. Нейропротекторные свойства снотворных средств при диффузном аксональном повреждении мозга // Сибирский медицинский журнал (Иркутск). 2016. Т. 145. № 6. С. 19-22.
12. Колударова Е.М., Тучик Е.С. Аспекты посмертной диагностики диффузного аксонального повреждения мозга // Вестник судебной медицины. 2019. Т. 8. № 3. С. 44-49.
13. Chen X.H., Siman R., Iwata A., Meaney D.F., Trojanowski J.Q., Smith D.H. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am. J. Pathol. 2004. vol. 165.no. 2. P. 357-371.
14. Netto C.B., Conte S., Leite M.C., Pires C., Martins T.L., Vidal P., Benfato M.S., Giugliani R., Gonçalves C.A. Serum S100B protein is increased in fasting rats. Arch. Med. Res. 2006. vol. 37. no. 5. P. 683–686.
15. Hostiuc S., Pirici D., Negoi I., Ion D.A., Ceausu M. Detection of diffuse axonal injury in forensic pathology. Romanian Journal of Legal Medicine. 2014. vol. 22. no. 3. P. 145-152.
16. Adams J.H., Graham D.I., Murray L.S., Scott G. Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann. Neurol. 1982. vol. 12. no. 6. P. 557-563.
17. Strich S.J. Diffuse degeneration of the cerebral white matter in severe dementia following head injury. J. Neurol.NeurosurgPsychiat. 1956. vol. 19. no. 69. P. 163-185.
18. Holbourn A.H. Mechanics of Head Injury. Lancet. 1943. vol. 2. no. 242. P. 438-441.
19. Pudenz R.H., SheldenC.H. Craniel trauma and brain movement. J. Neurosurg. 1946. vol. 3. P. 487-505.
20. Пашинян Г.А., Касумова С.Ю., Добровольский Г.Ф., Ромодановский П.О. Патоморфология и экспертная оценка повреждений головного мозга при черепно-мозговой травме. Москва, Ижевск.: Изд-во «Экспертиза». 1994. 134 с.
21. Gennarelli T.A., Thibault L.E., Adams J.H., Graham D.I., Thompson C.J., Marcincin R.P. Diffuse axonal injury and traumatic coma in the primate Ann. Neurol. 1982. vol. 12. no. 6. P. 564-574.
22. Povlishock J.T., Christman C.W., The Pathobiology of Traumatically Induced Axonal Injury in Animals and Humans: A Review of Current Thoughts. Journal of neurotrauma. 1995. vol. 12. no. 4. P. 555-564.
23. Dolinak D., Matshes E.W., Lew E.O. Forensic Pathology: Principles and Practice (1stedn). USA: Elsevier Academic Press. 2005. P. 442-446.
24. Adams J.H., Doyle D., Graham D.I., Lawrence A.E., McLellan D.R.. Diffuse axonal injury in head injuries caused by a fall. Lancet. 1984. vol. 324. no.8417. P. 1420-1422.
25. Graham D.I., Clark J.C., Adams J.H., Gennarelli T.A. Diffuse axonal injury caused by assault. J.Clin.Pathol. 1992. vol. 45. no. 9. P. 840–841.
26. Niess C, Grauel U, Toennes S.W., Bratzke H. Incidence of axonal injury in human brain tissue. ActaNeuropathol. 2002. vol. 104. no. 1. P. 79-84.
27. Shkrum M.J., Ramsay D.A. Forensic Pathology of Trauma: Common Problems for the Pathologists. (1stedn). Totowa New Jersey: Humana Press. 2007. P. 558-565.
28. Ромодановский П.О. Некоторые аспекты диффузного аксонального повреждения мозга при травме головы // Судебно-медицинская экспертиза. 2013. №3. С. 18-20.
29. Davceva N., Basheska N., Balazic J.DiffuseAxonalInjury – ADistinctClinicopathologicalEntity in Closed Head Injuries. J. Forensic Med Pathol. 2015. vol. 36. no. 3. P. 127-133.
30. Peerless S.J., Rewcastle N.B. Shear Injuries of the Brain. The Canadian Medical Association. 1967. vol. 96. no. 10. P. 577-582.
31. Kaur B., Rutty G.N., Timperley W.R. The possible role of hypoxia in the formation of axonal bulbs. J. Clin. Pathol. 1999. vol. 52. no. 3. P. 203–209.
32. Papa L., Robinson G. Use of biomarkers for diagnosis and management of traumatic brain injury patients.Expert Opinion on Medical Diagnostics. 2008. vol. 2. no. 8. P. 937-945.
33. Frati A.,Cerretani D., Fiaschi A.I.,Frati P., Gatto V.,Russa R.,Pesce A., Pinchi E., Santurro A., Fraschetti F.,Fineschi V. Diffuse Axonal Injury and Oxidative Stress: A Comprehensive Review. Int. J. Mol. Sci. 2017. vol. 18. no. 2. P. 1-20.
34. Lambri M., Djurovic V., Kibble M., Cairns N., Al-Sarraj S. Specificity and sensitivity of beta APP in head injury. Clin. Neuropathol. 2001. vol. 20. № 6. P. 263–271.
35. Reichard R.R., Smith C., Graham D.I. The significance of beta-APP immunoreactivity in forensic practice. Neuropathol. Appl. Neurobiol. 2005. vol. 31. no. 3. P. 304–313.
36. Beaudeux J., Dequen L., Foglietti M.Pathophysiologic aspects of S-100beta protein: a new biological marker of brain pathology. Ann. de biologie Clinique. 1999. vol. 57. no. 3. P. 261-272.
37. Isgro A., Bottoni P., Scatena R. NeuronSpecific Enolase as a Biomarker: Biochemical and Clinical Aspects. Adv. Exp. Med. Biol. 2015. vol. 867. P. 125-143.
38. Thelin E.P., Nelson D.W., Bellander B.M. Utilityofneuron-specificenolaseintraumaticbraininjury;relationstoS100Blevels,outcome,andextracranialinjuryseverity.CriticalCare.2016.vol. 20.no. 1. P. 1-15.
39. Papa L., Lewis L.M., Falk J.L., Zhang Z., Silvestri S., Giordano P., Brophy G.M., Demery J.A., Dixit N.K., Ferguson I., Liu M.C., Mo J., Akinyi L., Schmid K., Mondello S., Robertson C.S., Tortella F.C., Hayes R.L., Wang K.K. Elevated levels of serum glial fibrillary acidic protein breakdown products in mild and moderate traumatic brain injury are associated with intracranial lesions and neurosurgical intervention. Annals of Emergency Medicine. 2012. vol. 59. no. 6. P. 471-483.
40. Краснов А.В. Астроцитарные белки головного мозга: структура, функции, клиническое значение // Неврологический журнал. 2012. №17. С. 37-42.
41. Mondello S., Robicsek S.A., Gabrielli A., Brophy G. M., Papa L., Tepas J., Robertson G., Buki A., Scharf D., Mo J., Akinyi L., Muller U., Wang K.K.W., Hayes R. L. αII-spectrin breakdown products (SBDPs): diagnosis and outcome in severe traumatic brain injury patients. Journal of Neurotrauma. 2011. vol. 27. no. 7. P. 1203-1213.
42. Yokobori S., Hosein K., Burks S., Sharma I., Gajavelli S., Bullock R. Biomarkers for the Clinical Differential Diagnosis in Traumatic Brain Injury-A Systematic Review. CNS Neuroscience & Therapeutics. 2013. vol. 19.no. 8. Р. 556-565.
Диффузное аксональное повреждение (ДАП) как отдельная группа тяжелой черепно-мозговой травмы (ЧМТ) на протяжении многих лет привлекает внимание исследователей разных клинических специальностей [1-3]. Причиной этому отчасти служит отсутствие специфических критериев диагностики данной патологии, лабораторных маркеров этого патологического процесса. Меняется и подход к оценке тяжести и диагностики ДАП, использование шкалы комы Глазго и компьютерной томографии головного мозга в последнее время получили широкое внедрение в клиническую практику [4-6]. Появились работы по созданию алгоритмов формирования общей картины повреждений вещества головного мозга при ДАП с использованием программного обеспечения и вычислительной техники [7; 8]. Ведутся поиски различных вариантов фармакотерапии этой тяжелой формы травматического повреждения вещества головного мозга [9-11]. Несоответствие скудных морфологических проявлений и тяжести клинического состояния осложняет использование этого вида ЧМТ в практике судебно-медицинского эксперта, несмотря на достаточно изученный механизм травмы, приводящий к разрыву аксонов [12]. Пациентам с субдуральными, эпидуральными гематомами и другими формами ЧМТ диагноз ДАП не выставляется, несмотря на возможное повреждение аксональных трактов в этих случаях.
В настоящее время значительное количество работ, посвященных поиску различных биомаркеров-индикаторовДАП,в большинстве своем выполнены на животных,поэтомуих результаты при экстраполяции для применения в клинической практике необходимо интерпретировать с осторожностью [13; 14].
Следует отметить, что основная масса публикаций зарубежных и отечественных научных работ по этой тематике приходится на конец XX – начало XXI века. Были изучены клинические проявления ДАП, механизм травмы, макро- и микроскопическая картина при разных степенях тяжести и на различных сроках после получения травмы, начались исследования биомаркеров, позволяющих диагностировать травму головы, в том числе и повреждения аксонов. В последнее десятилетие интенсивность исследований по проблеме ДАП существенно снизилась. Появляющиеся единичные, в основном зарубежные, публикации, посвященные выявлению маркеров – индикаторов повреждения белого вещества головного мозга, служат целям клинической диагностики ДАП.
Чаще всего ДАП встречается при дорожно-транспортных происшествиях [15]. Поэтому в разрезе судебно-медицинских экспертных исследований проблема ДАП остается не до конца решенной. И принципиальным является не диагностика ДАП как основной или единственной составляющей черепно-мозговой травмы в рамках вопроса о причине смерти, а необходимость верифицировать ДАП при травме головы с переломами черепа, внутричерепными гематомами, ушибами мозга. К сожалению, в экспертной практике при наличии указанных тяжелых ЧМТ этому вопросу уделяется недостаточное внимание, при том что решение этого вопроса позволит экспертам более детально раскрыть механизм повреждений мозгового вещества и конкретизировать условия и этапытравмирования, что часто является ключевым при производстве таких экспертиз.Условия образования ДАП при различных механизмах травмы головы, скудность морфологической картины повреждения мозговой ткани при выраженной тяжести клинического течения посттравматического периода практически с момента травмы определяют необходимость в проведении углубленного анализа сведений и взглядов на ДАП с судебно-медицинских позиций для выработки алгоритма постановки диагноза при данной патологии.
Цель исследования: провести анализ зарубежных и отечественных публикацийпо проблеме диагностики ДАП в контексте возможности комплексного использования различных методик и биомаркеровв судебно-медицинских экспертных целях.
Материал и методы исследования.При подготовке публикации авторы использовали интернет-ресурсы: научная электронная библиотека (elibrary), SciVerse (ScienceDirect), Scopus, PubMed, GoogleScholar, BioMedSearch и Discover. Поиск публикаций осуществлялся по ключевым словам: диффузное аксональное повреждение, механизм ДАП, клиника ДАП, патоморфологическая картина при ДАП, гистологическая картина при ДАП, биомаркеры ДАП, diffuseaxonalinjury, themechanismoftheDAI, theDAIsymptoms, pathomorphologicalchangesintheDAI, histologicalchangesintheDAI, theDAIbiomarkers.После подборки научных публикаций описательным методом проводили обобщение и анализ информации о механизмах возникновения, диагностических критериях, клинических формах, вариантах течения, морфологической картине и биомаркерах ДАП.
Результаты исследования и их обсуждение.Термин «диффузное аксональное повреждение» был предложен в 1982 годуJ.H.Adamsс соавт. [16]. Они же описали характерные изменения в головном мозге при ДАП в виде местных повреждений, расположенных в мозолистом теле и (или) в дорсолатеральных квадрантах ростральных отделов мозга, а также диффузные изменения аксонов.
Однако первые описания дегенерации белого вещества головного мозга появились еще в 1940-х годах. Пионером в исследовании и описании этой патологии былаSabinaStrich [17], которая в 1956 году подробно описала морфологическую картину ЧМТ погибших после длительного пребывания в посттравматическом вегетативном состоянии с грубыми неврологическими нарушениями. Макроскопические признаки ЧМТ отсутствовали (за исключением одного пострадавшего с переломом костей черепа), что не соответствовало тяжести состояния, однако в ходе гистологического исследования была выявлена диффузная дегенерация белого вещества головного мозга, причиной которойявилось первичное физическое повреждение аксонов в момент травмы. Свои выводы S.Strich основывала на теоретических и экспериментальных работах A. Holbourn [18] и R.Pudenz, C.Shelden [19].
A.Holbourn, используя желатиновую модель головного мозга, воспроизводил вращательные движения в различных плоскостях (сагиттальной, вертикальной, горизонтальной). Он пришел к выводу, что формирующиеся в результате ускорения вращения«сдвиговые деформации»являются ведущим фактором, который вызывает разрыв синапсов, нервных волокон и кровеносных сосудов головного мозга. Важную рольвозникновения этого феномена авторотводитнесжимаемостии низкой ригидности вещества головного мозга при высокой ригидности черепа.
R.Pudenz,C.Sheldenзаменяли у приматов часть костей черепа прозрачной пластинкой инаносили удары с помощью специального аппарата на основе сжатого воздуха. В ходе исследования было установлено, что в момент удара мозг совершает вращательные и скользящие движения, направление которых зависит от точкиприложения силы. Движения мозговых полушарий наблюдались преимущественно в сагиттальной плоскости. Было отмечено, что удары в лобно-затылочном направленииприводят к смещению мозговых структур в сагиттальной плоскости, а на перемещение мозга при травматическом воздействии существенно влияла подвижность головы. Максимальные изменения отмечались при нефиксированной голове.
В то же времяГ.А.Пашинянс соавторами в монографии «Патоморфология и экспертная оценка повреждений головного мозга при черепно-мозговой травме» [20] утверждали, что повреждение чаще всего затрагивает плоскости между тканями различной плотности (соединение серого и белого вещества) и центр вращения мозга (ростральный отдел ствола). Локализация повреждения аксонов в стволе объясняется, прежде всего, тем, что головной мозг не фиксирован, поэтому при травматическом воздействии происходит некоторое его смещение. В свою очередь спинной мозг фиксирован корешками, что делает его устойчивым при вращении. На границе головного и спинного мозга располагается ствол, и это объясняет его преимущественное повреждение.
T.Gennarelliс соавт. [21] установили, что движение головы во фронтальной плоскости чаще приводит к серьезным аксональным повреждениям, тогда как движение ее в сагиттальной плоскости приводит к мягкому, в крайнем случае умеренному повреждению аксонов.
Несмотря на то что все вышеуказанные исследователи говорили о мгновенном разрыве аксонов под воздействием травмы, более поздниеработыJ. Povlishockс соавт. [22], как с участием экспериментальных животных, так и с использованием данных, полученных при аутопсии потерпевших с ЧМТ, показали, что патогенез травматически индуцированного аксонального повреждения является более сложным, чем первоначально предполагалось. Во многих случаях наблюдался каскад интрааксональных изменений, приводящий к прогрессивному набуханию аксонов и последующему разрыву в период с 12 до 24 часов.D.Dolinak с соавт.[23] с помощью гистохимического и других гистологических методов выяснили, что в момент травмы аксоны не рассекаются. Растяжение аксона приводит к его функциональному повреждению, которое заключается в травме микротрубочек и структуры нейрофиламентов. Цепь химических реакций в функционально поврежденном аксоне приводит к его постепенному отеку и разъединению. В частности, увеличивается проницаемость аксолеммы, и приток кальция запускает кальций-зависимые протеазы. Этот процесс еще сильнее повреждает аксон, что приводит к накоплению аксонального содержимого в месте разрушения. Сначала это приводит к набуханию (аксональные утолщения), а затем к полному разъединению и формированию аксональных шаров.
Наибольшие предпосылки для реализации указанных механизмов имеются при высокоскоростных дорожно-транспортных происшествиях, что определяет значимость изучения ДАП для реконструкции событий при проведении судебно-медицинских экспертиз. Однако имеются данные, свидетельствующие об ином механизме получения данного повреждения: в результате падения с большой высоты; повреждения, полученные при разбойном нападении [24; 25]. C. Niessс соавт.[26] приводят случай повреждения аксонов в результате передозировки наркотическими препаратами, когда при вскрытии была диагностирована субдуральная гематома, при отсутствии переломов костей черепа. В этом случае, по мнению авторов,причиной повреждения аксонов явилась гипоксия при действии на дыхательный центр наркотических веществ. Хотя эти данные представляются весьма сомнительными, так как авторы не приводят сведения катамнеза, исключающие травму, однако исследования, опровергающие указанный механизм образования повреждений аксонов,отсутствуют.
M.J. Shkrum, D.A.Ramsay [27] считают нехарактерным обнаружение контактных повреждений при ДАП, но отмечают, что в некоторых случаяхвстречаются повреждения волосистой части головы, трещиныкостей черепа, субдуральные и субарахноидальные кровоизлияния или ушиб коры головного мозга. По мнению авторов, для ДАП более характерны очаговые кровоизлияния, расположенные в глубоких отделах белого вещества головного мозга (мозолистое тело, внутренняя капсула, верхние ножки мозжечка).При тяжелой степени ДАП взаимное движение между различными участками мозга приводит к разрыву аксонов вместе с мелкими кровеносными сосудами что, в свою очередь, ведет к множественным петехиальным кровоизлияниям в головном мозге, преимущественно локализующимся в белом веществе лобной доли. Это явление носит название «диффузный васкулярный ушиб», отражает тяжесть диффузной травмы мозга и имеет короткий период выживаемости (от нескольких минут до нескольких часов).
Изменение патоморфологической картиныв головном мозгев зависимости от срока, прошедшего после получения травмы, нашло отражение в работе П.О.Ромодановского [28]. Так, при смерти в течение первыхтрех суток отмечается набухание мозга, мелкоочаговые и очаговые кровоизлияния в мозолистом теле, базальных ядрах, семиовальном центре, реже в стволе. При смерти спустя 1 неделю после травмы появляются признаки давности геморрагий в глубинные отделы.Спустя месяц обнаруживаютсябурые кисты в области первичного повреждения белого вещества. А при сроке более 3 месяцев обнаруживаются признаки атрофии мозга, вентрикуломегалии, расширение субарахноидальных пространств.
Для гистологической верификации ДАП автор предлагает комплекс методов окраски, которыми следует пользоваться с учетом сроков посттравматического периода. Основными из них являются методики, позволяющие обнаружить изменения осевых цилиндров (импрегнация серебром по Бильшовскому или Глису), миелиновых оболочек (импрегнация осмием по Марки для выявления ранней демиелинизации, окраска по Шпильмейеру для обнаружения поздней демиелинизации).
N.Davceva с соавт.[29] выделяют два основных признака, на основании которых можно поставить диагноз ДАП. Первый — это наличие диффузного повреждения аксонов белого вещества, и второй — широкая распространенность данного повреждения таким образом, что хотя бы одна поврежденная область располагается выше и одна ниже Tentorium cerebelli, обращая особое внимание на аксональные тракты (мозолистое тело, внутренняя капсула).
Исследования, проведенные S.J.Peerless, N.B.Rewcastle [30], показали, что для ДАП характерным является обнаружение поврежденных аксонов – аксональных утолщений или аксональных шаров – при микроскопическом исследовании. Аксональные утолщения обусловлены удлинением аксонов без разрыва и характеризуются участками утолщения аксонов, разделенными истонченными участками. Аксональные шары (шары Марки, терминальные, ретрагированные шары) встречаются в ранние сроки, после травмы (впервые их можно увидеть в течение двух часов после травмы, а максимального развития они достигают спустя 4 часа). Механизм их возникновения авторы объясняютвытеканием аксоплазмы из обоих концов поврежденного участка нервного волокна при разрыве с формированием булавовидных утолщений. На дистальном сегменте аксональные шары регистрируются лишь в течение 5-7 дней, тогда как на проксимальном сегменте регулярно встречаются в течение многих месяцев и даже лет после травмы, в том случае, если тело клетки выживает. Однако,по мнению B.Kaur с соавт.[31], аксональные шары не являются строго специфичными для травматического генеза повреждения аксонов. Их появление возможно, например, при гипоксии и отсутствии травмы головы.
В работе Г.А. Пашиняна с соавт. [20] отмечается, что с 5-го дня после травмы определялись участки ранней дегенерации белого вещества в областях первичных повреждений. Со второй недели выявлялись выраженные дегенеративные изменения в зонах повреждений и начальные признаки вторичной посттравматической дегенерации с образованием зернистых шаров. При сроке более двух недель появлялись выраженные дегенеративные изменения нервной ткани как в зонах повреждения белого вещества, так и на отдалении. При сроке более месяца в зонах первичных повреждений определялась демиелинизация аксональных структур. При переживании травмы более 3 месяцев наблюдалась диффузная вторичная дегенерация нервной ткани, спинного мозга и в периферической нервной системе.
Помимо вышеуказанных, высокоинформативным методом диагностики (прижизненным и посмертным) является определение биомаркеров повреждения головного мозга. Исходя из анализа исследований, посвященных биомаркерам, проведенного L.Papa, G. Robinson[32], можно сделать вывод, что специфических маркеров ДАП до сих пор не найдено, а результаты многочисленных исследований противоречивы. Но, несмотря на это, исследователи утверждают, что простота использования, точная ранняя диагностика как прижизненно, так и посмертно диктует необходимость в продолжении исследований в отношении биомаркеров повреждения мозга.
X.H.Chen с соавт. [13] и A. Fratiс соавт. [33] предлагают «золотым стандартом» диагностики ДАП считать выявление β-APP белков. Предшественники бета-амилоида(amyloidprecursorprotein, АРР) — это трансмембранные белки, в больших количествах локализующиеся в синаптических мембранах.Их функция пока остается неизвестной. В результате их протеолиза образуются амилоид-β1–40 и амилоид-β1–42. После ЧМТ продукты протеолизаAPP и активированная капсаза-3, а также другие протеолитические ферменты обнаруживаются в аксонах и теле нейрона. Иммуногистохимическим (ИГХ) методом, с помощью антител к β-APP, идентифицируют пораженные аксоны уже через 2-3 часа после травмы. Они остаются видимыми при ИГХ-исследовании длительное время -до 6 месяцев. Следует отметить, что,по данным M. Lambriс соавт. [34] и R. Reichardс соавт.[35], имеются определенные морфологические закономерности распределения иммунореактивных аксонов, позволяющие на микроскопическом уровне отличить травматическое повреждение аксонов от других видов повреждения (гипоксия и т.д.).
J. Beaudeux с соавт. [36] описывает другой примербиомаркера повреждения мозга — белок S-100. Это кальций-связывающий белок, секретируемый преимущественно глиальными клетками. Структурное повреждение этих клеток вызывает утечку этого белка во внеклеточное пространство, затем в спинномозговую жидкость и его дальнейшее поступление в кровоток. Таким образом, повышение концентрации белка S-100 было предложено в качестве индекса повреждения ткани головного мозга.В настоящее времяобсуждается возможность синтеза данного белка другими тканями (например, адипоцитами) [14].
Были обнаружены предиктивные способности нейрон-специфической енолазы – внутриклеточного фермента центральной нервной системы, локализующегося в клетках нейроэктодермального происхождения. Это делает его высокоспецифичным маркером повреждения таких клеток. Однако чаще его используют в диагностике различных злокачественных новообразований [37].
E. Thelinс соавт.[38] обратили внимание, что изменение концентрации белка S-100 было лучшим предиктором травмы головного мозга по сравнению с изменением концентрации нейрон-специфической енолазы. Несмотря на это, оба биомаркера обладают предиктивной способностью независимо друг от друга; способности нейрон-специфической енолазы ограничены при наличии белка S-100, т.к. между ними имеется высокая зависимость. Более того, нейрон-специфическая енолаза в большей степени коррелирует с мультитравмой, однако эффект экстрацеребральной травмы, по-видимому, ограничен первыми 48 часами. Из вышесказанного можно сделать вывод, что хотя оба биомаркера независимо коррелируют с ЧМТ, белокS-100 является более точным предиктором травмы головного мозга и, возможно, более клинически полезным биомаркером, чем нейрон-специфическая енолаза.
В качестве биомаркера повреждения L.Papaс соавт. [39] в своих исследованиях рассматривают глиальный фибриллярный кислый протеин (GFAP). GFAP – это член семейства белков цитоскелета, промежуточный филамент в зрелых астроцитах. Появление его в сыворотке крови является предиктором поражения различных структур мозгового вещества. Помимо этого, стабильно высокий уровень GFAP в течение 2 дней после травмы значительно повышает риск летального исхода у пациентов.
Еще одним предиктором повреждения ткани мозга является альфа-II-спектрин. Альфа-II-спектрин – это компонент цитоскелета нейронов коры головного мозга, субстрат для кальций-активируемых цистеиновыхпротеиназ (калпаин, капсаза-3). Обнаруживается также в пресинаптических окончаниях или в субаксолемномкомпартменте аксонов. Протеиназы являются основными факторами, вызывающими некротические и апоптотические процессы в клетках при ишемии и травме головного мозга, соответственно. Они активируются после травмы и расщепляют спектрин, в результате в цереброспинальной жидкости и тканях мозга значительно повышается концентрация спектрина и продуктов его распада (SBDPs145 – производное калпаина, SBDPs120 – производное каспазы-3). Средняя концентрация SBDPs145 достигает своего пика рано (в течение 6часов после травмы) и снижается медленно (в течение 7 дней). SBDPs120 показывает устойчивый подъем, который сохраняется в течение как минимум 7 дней, тогда как пик концентрации приходится на 5-е сутки. На 2-3-е сутки после ЧМТ уровень альфа-II-спектрина достигает максимума [40; 41].
Рассматривается возможность применения расщепленного тау-белка (MAP-tau) в качестве биомаркера повреждения мозга.Он представляет собой фосфопротеин, ассоциированный с микротрубочками. Чаще всего встречается в нейронах ЦНС, локализуется в аксональном компартменте. В результате повреждения аксонов MAP-tau подвергается протеолизу и получает доступ к спинномозговой жидкости и сыворотке крови. В течение первых 24 часов уровень MAP-tau достигает своего пика, затем постепенно снижается в течение 7 суток [42].
Другие потенциальные биомаркеры при ЧМТ: убиквинтин С-терминальная гидролаза L1 (ubiquitin C-terminalhydrolase, UCH-1), воспалительные цитокины и др., изучены недостаточно и не являются диагностически значимыми.
Заключение
Таким образом, проведенный анализ публикаций результатов исследований ДАП показал отсутствие сформированного, научнообоснованного алгоритма применения клинико-морфологических критериев судебно-медицинской диагностики данной патологии в случаях тяжелой черепно-мозговой травмы. Это свидетельствует о необходимости проведения исследований для поиска комплексного решения данного вопроса с применением различныхметодов исследования (анализ клинической картины, морфологических, гистологических изменений и биомаркеров) в случаях с подозрением на ДАП в судебно-медицинской практике.
Библиографическая ссылка
Ефимов А.А., Савенкова Е.Н., Алексеев Ю.Д., Райкова К.А., Коротина О.С., Корсак В.О. ДИФФУЗНОЕ АКСОНАЛЬНОЕ ПОВРЕЖДЕНИЕ МОЗГА С СУДЕБНО-МЕДИЦИНСКИХ ПОЗИЦИЙ // Современные проблемы науки и образования. – 2020. – № 5.
;
URL: https://science-education.ru/ru/article/view?id=30140 (дата обращения: 05.04.2023).
Предлагаем вашему вниманию журналы, издающиеся в издательстве «Академия Естествознания»
(Высокий импакт-фактор РИНЦ, тематика журналов охватывает все научные направления)
From Wikipedia, the free encyclopedia
| Diffuse axonal injury | |
|---|---|
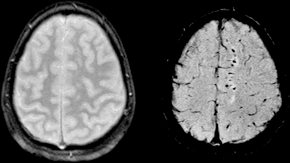 |
|
| Two MRI images of a patient with diffuse axonal injury resulting from trauma, at 1.5 tesla field strength. Left: conventional gradient recalled echo (GRE). Right: Susceptibility weighted image (SWI). | |
| Specialty | Neurology |
Diffuse axonal injury (DAI) is a brain injury in which scattered lesions occur over a widespread area in white matter tracts as well as grey matter.[1][2][3][4][5][6][7] DAI is one of the most common and devastating types of traumatic brain injury[8] and is a major cause of unconsciousness and persistent vegetative state after severe head trauma.[9] It occurs in about half of all cases of severe head trauma and may be the primary damage that occurs in concussion. The outcome is frequently coma, with over 90% of patients with severe DAI never regaining consciousness.[9] Those who awaken from the coma often remain significantly impaired.[10]
DAI can occur across the spectrum of traumatic brain injury (TBI) severity, wherein the burden of injury increases from mild to severe.[11][12] Concussion may be a milder type of diffuse axonal injury.[12][13]
Mechanism[edit]
DAI is the result of traumatic shearing forces that occur when the head is rapidly accelerated or decelerated, as may occur in car accidents, falls, and assaults.[14] Vehicle accidents are the most frequent cause of DAI; it can also occur as the result of child abuse[15] such as in shaken baby syndrome.[16]
Immediate disconnection of axons may be observed in severe brain injury, but the major damage of DAI is delayed secondary axon disconnections, slowly developed over an extended time course.[2] Tracts of axons, which appear white due to myelination, are referred to as white matter. Lesions in both grey and white matter are found in postmortem brains in CT and MRI exams.[9]
Besides mechanical breakage of the axonal cytoskeleton, DAI pathology also includes secondary physiological changes, such as interrupted axonal transport, progressive swelling and degeneration.[17] Recent studies have linked these changes to twisting and misalignment of broken axon microtubules, as well as tau protein and amyloid precursor protein (APP) deposition.[17][18]
Characteristics[edit]
Lesions typically are found in the white matter of brains injured by DAI; these lesions vary in size from about 1–15 mm and are distributed in a characteristic pattern.[9] DAI most commonly affects white matter in areas including the brain stem, the corpus callosum, and the cerebral hemispheres.
The lobes of the brain most likely to be injured are the frontal and temporal lobes.[19] Other common locations for DAI include the white matter in the cerebral cortex, the superior cerebral peduncles,[16] basal ganglia, thalamus, and deep hemispheric nuclei.[clarification needed][20] These areas may be more easily damaged because of the difference in density between them and the other regions of the brain.[20]
Histological characteristics[edit]
DAI is characterized by axonal separation, in which the axon is torn at the site of stretch and the part distal to the tear degrades by a process known as Wallerian degeneration. While it was once thought that the main cause of axonal separation was tearing due to mechanical forces during the trauma event, it is now understood that axons are not typically torn upon impact; rather, secondary biochemical cascades, which occur in response to the primary injury (which occurs as the result of mechanical forces at the moment of trauma) and take place hours to days after the initial injury, are largely responsible for the damage to axons.[21][22][23]
Though the processes involved in secondary brain injury are still poorly understood, it is now accepted that stretching of axons during injury causes physical disruption to and proteolytic degradation of the cytoskeleton.[24] It also opens sodium channels in the axolemma, which causes voltage-gated calcium channels to open and Ca2+ to flow into the cell.[24] The intracellular presence of Ca2+ triggers several different pathways, including activating phospholipases and proteolytic enzymes damaging mitochondria and the cytoskeleton, and activating secondary messengers, which can lead to separation of the axon and death of the cell.[21]
Cytoskeleton disruption[edit]

Immunoreactive axonal profiles are observed as either granular (B, G, H) or more elongated, fusiform (F) swellings in the corpus callosum and the brain stem (H) at 24h post traumatic brain injury. Example of APP immunoreactive neurons (arrow heads) observed in the cortex underneath the impact site (E, G). No APP staining was observed in healthy control animals (D).[23]
Axons are normally elastic, but when rapidly stretched they become brittle, and the axonal cytoskeleton can be broken. Misalignment of cytoskeletal elements after stretch injury can lead to tearing of the axon and death of the neuron. Axonal transport continues up to the point of the break in the cytoskeleton, but no further, leading to a buildup of transport products and local swelling at that point.[25] When this swelling becomes large enough, it can tear the axon at the site of the cytoskeleton break, causing it to draw back toward the cell body and form a bulb.[11] This bulb is called a «retraction ball», the histological hallmark of diffuse axonal injury.[9]
When the axon is torn, Wallerian degeneration, in which the part of the axon distal to the break degrades, takes place within one to two days after injury.[26] The axolemma disintegrates,[26] myelin breaks down and begins to detach from the cell in an anterograde direction (from the body of the cell toward the end of the axon),[27] and nearby cells begin phagocytic activity, engulfing the cellular debris.[28]
Calcium influx[edit]
While sometimes only the cytoskeleton is disturbed, frequently disruption of the axolemma occurs as well, causing the influx of Ca2+ ions into the cell and unleashing a variety of degradational processes.[26][29] An increase in Ca2+ and Na+ levels and a drop in K+ levels are found within the axon immediately after injury.[21][26] Possible routes of Ca2+ entry include sodium channels, pores formed in the membrane during stretch, and failure of ATP-dependent transporters due to mechanical blockage or lack of available metabolic energy.[21] High levels of intracellular Ca2+, the major cause of post-injury cell damage,[30] destroy mitochondria,[11] and trigger phospholipases and proteolytic enzymes that damage Na+ channels and degrade or alter the cytoskeleton and the axoplasm.[31][26] Excess Ca2+ can also lead to damage to the blood–brain barrier and swelling of the brain.[30]
One of the proteins activated by the presence of calcium in the cell is calpain, a Ca2+-dependent non-lysosomal protease.[31] About 15 minutes to half an hour after the onset of injury, a process called calpain-mediated spectrin proteolysis, or CMSP, begins to occur.[32] Calpain breaks down a molecule called spectrin, which holds the membrane onto the cytoskeleton, causing the formation of blebs and the breakdown of the cytoskeleton and the membrane, and ultimately the death of the cell.[31][32] Other molecules that can be degraded by calpains are microtubule subunits, microtubule-associated proteins, and neurofilaments.[31]
Generally occurring one to six hours into the process of post-stretch injury, the presence of calcium in the cell initiates the caspase cascade, a process in cell injury that usually leads to apoptosis, or «programmed cell death».[32]
Mitochondria, dendrites, and parts of the cytoskeleton damaged in the injury have a limited ability to heal and regenerate, a process which occurs over two or more weeks.[33] After the injury, astrocytes can shrink, causing parts of the brain to atrophy.[9]
Diagnosis[edit]

Diffuse axonal injury after a motorcycle accident. MRI after 3 days: on T1-weighted images the injury is barely visible. On the FLAIR, DWI and T2*-weighted images a small bleed is identifiable.
DAI is difficult to detect since it does not show up well on CT scans or with other macroscopic imaging techniques, though it shows up microscopically.[9] However, there are characteristics typical of DAI that may or may not show up on a CT scan. Diffuse injury has more microscopic injury than macroscopic injury and is difficult to detect with CT and MRI, but its presence can be inferred when small bleeds are visible in the corpus callosum or the cerebral cortex.[34] MRI is more useful than CT for detecting characteristics of diffuse axonal injury in the subacute and chronic time frames.[35] Newer studies such as Diffusion Tensor Imaging are able to demonstrate the degree of white matter fiber tract injury even when the standard MRI is negative. Since axonal damage in DAI is largely a result of secondary biochemical cascades, it has a delayed onset, so a person with DAI who initially appears well may deteriorate later. Thus injury is frequently more severe than is realized, and medical professionals should suspect DAI in any patients whose CT scans appear normal but who have symptoms like unconsciousness.[9]
MRI is more sensitive than CT scans, but is still liable to false negatives because DAI is identified by looking for signs of edema, which may not always be present.[33]
DAI is classified into grades based on severity of the injury. In Grade I, widespread axonal damage is present but no focal abnormalities are seen. In Grade II, damage found in Grade I is present in addition to focal abnormalities, especially in the corpus callosum. Grade III damage encompasses both Grades I and II plus rostral brain stem injury and often tears in the tissue.[36]
Treatment[edit]
DAI currently lacks specific treatment beyond that for any type of head injury, which includes stabilizing the patient and trying to limit increases in intracranial pressure (ICP).
History[edit]
The idea of DAI first came about as a result of studies by Sabina Strich on lesions of the white matter of individuals who had sustained head trauma years before.[37] Strich first proposed the idea in 1956, calling it diffuse degeneration of white matter; however, the more concise term «diffuse axonal injury» came to be preferred.[38] Strich was researching the relationship between dementia and head trauma[37] and asserted in 1956 that DAI played an integral role in the eventual development of dementia due to head trauma.[15] The term DAI was introduced in the early 1980s.[39]
Notable examples[edit]
- Top Gear presenter Richard Hammond sustained a DAI as a result of the Vampire dragster crash in 2006.
- Champ Car World Series driver Roberto Guerrero suffered a DAI as a result of a crash during testing at the Indianapolis Motor Speedway in 1987.[40]
- Formula 1 driver Jules Bianchi suffered a DAI as a result of an accident at the 2014 Japanese Grand Prix[41] and died, without regaining consciousness, 9 months later on 17 July 2015.[42]
- Actor and audiobook narrator Frank Muller, who read Stephen King’s The Dark Tower, suffered a DAI in 2001 due to a motorcycle accident. He died in 2008.[43]
- NASCAR driver Adam Petty, grandson of seven time Cup Series champion Richard Petty, sustained a diffuse axonal injury secondary to a fatal basilar skull fracture in May 2000 at New Hampshire Motor Speedway during practice for the upcoming race.
See also[edit]
- Brain injury
- Axoplasmic transport
References[edit]
- ^ Strich SJ (August 1956). «Diffuse degeneration of the cerebral white matter in severe dementia following head injury». Journal of Neurology, Neurosurgery, and Psychiatry. 19 (3): 163–85. doi:10.1136/jnnp.19.3.163. PMC 497203. PMID 13357957.
- ^ a b Povlishock JT, Becker DP, Cheng CL, Vaughan GW (May 1983). «Axonal change in minor head injury». Journal of Neuropathology and Experimental Neurology. 42 (3): 225–42. doi:10.1097/00005072-198305000-00002. PMID 6188807. S2CID 24260379.
- ^ Adams JH (March 1982). «Diffuse axonal injury in non-missile head injury». Injury. 13 (5): 444–5. doi:10.1016/0020-1383(82)90105-X. PMID 7085064.
- ^ Christman CW, Grady MS, Walker SA, Holloway KL, Povlishock JT (April 1994). «Ultrastructural studies of diffuse axonal injury in humans». Journal of Neurotrauma. 11 (2): 173–86. doi:10.1089/neu.1994.11.173. PMID 7523685.
- ^ Povlishock JT, Christman CW (August 1995). «The pathobiology of traumatically induced axonal injury in animals and humans: a review of current thoughts». Journal of Neurotrauma. 12 (4): 555–64. doi:10.1089/neu.1995.12.555. PMID 8683606.
- ^ Vascak M, Jin X, Jacobs KM, Povlishock JT (May 2018). «Mild Traumatic Brain Injury Induces Structural and Functional Disconnection of Local Neocortical Inhibitory Networks via Parvalbumin Interneuron Diffuse Axonal Injury». Cerebral Cortex. 28 (5): 1625–1644. doi:10.1093/cercor/bhx058. PMC 5907353. PMID 28334184.
- ^ Smith DH, Hicks R, Povlishock JT (March 2013). «Therapy development for diffuse axonal injury». Journal of Neurotrauma. 30 (5): 307–23. doi:10.1089/neu.2012.2825. PMC 3627407. PMID 23252624.
- ^ Povlishock JT, Katz DI (January 2005). «Update of neuropathology and neurological recovery after traumatic brain injury». The Journal of Head Trauma Rehabilitation. 20 (1): 76–94. doi:10.1097/00001199-200501000-00008. PMID 15668572. S2CID 1094129.
- ^ a b c d e f g h Wasserman J. and Koenigsberg R.A. (2007). Diffuse axonal injury. Emedicine.com. Retrieved on 2008-01-26.
- ^ Vinas F.C. and Pilitsis J. (2006). Penetrating head trauma. Emedicine.com. Retrieved on 2008-01-14.
- ^ a b c Smith DH, Meaney DF (December 2000). «Axonal damage in traumatic brain injury». The Neuroscientist. 6 (6): 483–95. doi:10.1177/107385840000600611. S2CID 86550146.
- ^ a b Blumbergs PC, Scott G, Manavis J, Wainwright H, Simpson DA, McLean AJ (August 1995). «Topography of axonal injury as defined by amyloid precursor protein and the sector scoring method in mild and severe closed head injury». Journal of Neurotrauma. 12 (4): 565–72. doi:10.1089/neu.1995.12.565. PMID 8683607.
- ^ Bazarian JJ, Blyth B, Cimpello L (February 2006). «Bench to bedside: evidence for brain injury after concussion—looking beyond the computed tomography scan». Academic Emergency Medicine. 13 (2): 199–214. doi:10.1197/j.aem.2005.07.031. PMID 16436787.
- ^ Gennarelli TA (1993). «Mechanisms of brain injury». The Journal of Emergency Medicine. 11 Suppl 1: 5–11. PMID 8445204.
- ^ a b
Hardman JM, Manoukian A (May 2002). «Pathology of head trauma». Neuroimaging Clinics of North America. 12 (2): 175–87, vii. doi:10.1016/S1052-5149(02)00009-6. PMID 12391630. - ^ a b Smith D. and Greenwald B. 2003.Management and staging of traumatic brain injury. Emedicine.com. Retrieved through web archive on 17 January 2008.
- ^ a b Johnson VE, Stewart W, Smith DH (August 2013). «Axonal pathology in traumatic brain injury». Experimental Neurology. Special Issue: Axonal degeneration. 246: 35–43. doi:10.1016/j.expneurol.2012.01.013. PMC 3979341. PMID 22285252.
- ^ Tang-Schomer MD, Patel AR, Baas PW, Smith DH (May 2010). «Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration». FASEB Journal. 24 (5): 1401–10. doi:10.1096/fj.09-142844. PMC 2879950. PMID 20019243.
- ^ Boon R, de Montfor GJ (2002). «Brain injury». Learning Discoveries Psychological Services. Archived from the original on 2006-09-03. Retrieved 17 January 2008.
- ^ a b Singh J, Stock A (September 25, 2006). «Head Trauma». Emedicine.com. Retrieved 2008-01-17.
- ^ a b c d Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH (2001). «Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels». Journal of Neuroscience. 21 (6): 1923–1930. doi:10.1523/JNEUROSCI.21-06-01923.2001. PMC 6762603. PMID 11245677.
- ^ Arundine M, Aarts M, Lau A, Tymianski M (September 2004). «Vulnerability of central neurons to secondary insults after in vitro mechanical stretch». Journal of Neuroscience. 24 (37): 8106–23. doi:10.1523/JNEUROSCI.1362-04.2004. PMC 6729801. PMID 15371512.
- ^ a b Mouzon B, Chaytow H, Crynen G, Bachmeier C, Stewart J, Mullan M, Stewart W, Crawford F (December 2012). «Repetitive mild traumatic brain injury in a mouse model produces learning and memory deficits accompanied by histological changes» (PDF). Journal of Neurotrauma. 29 (18): 2761–2173. doi:10.1089/neu.2012.2498. PMID 22900595.
- ^ a b Iwata A, Stys PK, Wolf JA, Chen XH, Taylor AG, Meaney DF, Smith DH (2004). «Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors». The Journal of Neuroscience. 24 (19): 4605–4613. doi:10.1523/JNEUROSCI.0515-03.2004. PMC 6729402. PMID 15140932.
- ^ Staal JA, Dickson TC, Chung RS, Vickers JC (2007). «Cyclosporin-A treatment attenuates delayed cytoskeletal alterations and secondary axotomy following mild axonal stretch injury». Developmental Neurobiology. 67 (14): 1831–1842. doi:10.1002/dneu.20552. PMID 17702000. S2CID 19415197.
- ^ a b c d e LoPachin RM, Lehning EJ (1997). «Mechanism of calcium entry during axon injury and degeneration». Toxicology and Applied Pharmacology. 143 (2): 233–244. doi:10.1006/taap.1997.8106. PMID 9144441.
- ^ Cowie RJ, Stanton GB (2005). «Axoplasmic transport and neuronal responses to injury». Howard University College of Medicine. Archived from the original on 2005-10-29. Retrieved 2008-01-17.
- ^ Hughes PM, Wells GM, Perry VH, Brown MC, Miller KM (2002). «Comparison of matrix metalloproteinase expression during wallerian degeneration in the central and peripheral nervous systems». Neuroscience. 113 (2): 273–287. doi:10.1016/s0306-4522(02)00183-5. PMID 12127085. S2CID 37213275.
- ^ Povlishock JT, Pettus EH (1996). «Traumatically induced axonal damage: Evidence for enduring changes in axolemmal permeability with associated cytoskeletal change». Acta Neurochirurgica. 66: 81–86. doi:10.1007/978-3-7091-9465-2_15. ISBN 978-3-7091-9467-6. PMID 8780803.
- ^ a b Zhou F, Xiang Z, Feng WX, Zhen LX (2001). «Neuronal free Ca2+ and BBB permeability and ultrastructure in head injury with secondary insult». Journal of Clinical Neuroscience. 8 (6): 561–563. doi:10.1054/jocn.2001.0980. PMID 11683606. S2CID 43789581.
- ^ a b c d Castillo MR, Babson JR (1998). «Ca2+-dependent mechanisms of cell injury in cultured cortical neurons». Neuroscience. 86 (4): 1133–1144. doi:10.1016/s0306-4522(98)00070-0. PMID 9697120. S2CID 54228571.
- ^ a b c Büki A, Okonkwo DO, Wang KK, Povlishock JT (April 2000). «Cytochrome c release and caspase activation in traumatic axonal injury». primary. The Journal of Neuroscience. 20 (8): 2825–34. doi:10.1523/JNEUROSCI.20-08-02825.2000. PMC 6772193. PMID 10751434.
- ^ a b Corbo J, Tripathi P (2004). «Delayed presentation of diffuse axonal injury: A case report». Trauma. 44 (1): 57–60. doi:10.1016/j.annemergmed.2003.11.010. PMID 15226709.
- ^
Crooks CY, Zumsteg JM, Bell KR (November 2007). «Traumatic brain injury: a review of practice management and recent advances». Physical Medicine and Rehabilitation Clinics of North America. 18 (4): 681–710, vi. doi:10.1016/j.pmr.2007.06.005. PMID 17967360. - ^ Maas AI, Stocchetti N, Bullock R (August 2008). «Moderate and severe traumatic brain injury in adults». The Lancet. Neurology. 7 (8): 728–41. doi:10.1016/S1474-4422(08)70164-9. PMID 18635021. S2CID 14071224.
- ^ Lees-Haley PR, Green P, Rohling ML, Fox DD, Allen LM (August 2003). «The lesion(s) in traumatic brain injury: implications for clinical neuropsychology». Archives of Clinical Neuropsychology. 18 (6): 585–94. doi:10.1016/S0887-6177(02)00155-5. PMID 14591433.
- ^ a b
Pearce JM (2007). «Observations on concussion. A review». European Neurology. 59 (3–4): 113–9. doi:10.1159/000111872. PMID 18057896. S2CID 10245120. - ^
Gennarelli GA, Graham DI (2005). «Neuropathology». In Silver JM, McAllister TW, Yudofsky SC (eds.). Textbook Of Traumatic Brain Injury. Washington, DC: American Psychiatric Association. p. 34. ISBN 978-1-58562-105-7. Retrieved 2008-06-10. - ^ Granacher RP (2007). Traumatic Brain Injury: Methods for Clinical & Forensic Neuropsychiatric Assessment, Second Edition. Boca Raton: CRC. pp. 26–32. ISBN 978-0-8493-8138-6. Retrieved 2008-07-06.
- ^ «The story of Roberto Guerrero».
- ^ «Jules Bianchi: Family confirms Formula One driver sustained traumatic brain injury in Japanese GP crash». Retrieved 8 October 2014.
- ^ «F1 driver Jules Bianchi dies from crash injuries». BBC Sport. BBC. 2015-07-18. Retrieved 18 July 2015.
- ^ «Frank Muller, The Fight of his Life». 2006. Retrieved December 14, 2017.
External links[edit]
- Diffuse Axonal Injury MRI and CT Images
Диффузное аксональное повреждение головного мозга (ДАП) – это травма с поражением аксонов. При таком повреждении нарушается целостность аксонов, передающих нервные сигналы от клеточных тел к иннервируемым органам и другим нейронам.
Последствия черепно-мозговой травмы (ЧМТ) с диффузным повреждением аксонов очень серьезные. Человек практически сразу впадает в кому, которая грозит переходом в вегетативное состояние. Непосредственной причиной являются надрывы и разрывы аксонов – нейритов, представляющих собой длинные цилиндрические отростки нервных клеток. Помимо этого, в структурах мозга образуются мелкоочаговые кровоизлияния.
При ДАП-синдроме поражаются, как правило, следующие зоны:
- ствол головного мозга;
- белое вещество полушарий;
- мозолистое тело;
- перивентрикулярные зоны мозга.
Аксональное повреждение головного мозга наиболее часто встречается у детей, включая новорожденных, и молодежи. В детском возрасте неврологический дефицит более выражен, а кома более глубокая.
Причины
Синдром диффузного повреждения мозга возникает в результате травм, которые обусловлены боковым ускорением черепа. Для этого не требуется прямой удар по голове, поэтому переломы и видимые раны часто отсутствуют.
Если траектория головы в момент травмы вертикальная, повреждаются преимущественно сосуды с формированием гематом в мозговом веществе. Ускорение в угловой и боковой плоскости приводит к диффузному повреждению аксонов мозга.
Диффузное поражение вызывают травмы, полученные в ДТП, при падении с высоты и резком изменении внешнего давления (подводные погружения, взрывы, падение давления в барокамере, некорректное применение ИВЛ и пр.).
Перечисленные виды травм предполагают боковое ускорение головы с поворотами более подвижных полушарий мозга и перекручиванием более стабильных. Не исключен также сдвиг отдельных мозговых слоев или отделов, который может спровоцировать надрыв или разрыв аксонов и мелких капилляров. Достаточно незначительного смещения церебральных структур для нарушения целостности нейритов и сосудов.
![]()
Агишев Дамир Адгемович
врач – невролог
Запишитесь на онлайн-консультацию, если ваш близкий человек попал в беду и находится в коме. Наши врачи дистанционно – по телефону или видеосвязи – изучат историю болезни и расскажут, чем вы можете ему помочь. При необходимости порекомендуют профильную клинику. Консультации проводятся круглосуточно.
Online консультация
Симптомы
Типичным симптомом диффузного аксонального повреждения головного мозга является кома. Она может быть умеренной или глубокой, но всегда возникает в первые минуты после травмы.
В большинстве случаев кома переходит в вегетативное состояние, при котором надолго останавливается деятельность коры головного мозга. В среднем кома длится около недели.
Для диффузии головного мозга характерны хаотичные телодвижения пациента, возникающие внезапно на фоне общей обездвиженности. Снижается или пропадает зрачковый рефлекс, отмечается разный диаметр зрачков (анизокория), которые часто находятся не на одной горизонтальной линии.
У пациентов с диффузным аксональным поражением мозга нарушается частота и ритм дыхания, твердеют затылочные мышцы. Отмечается симптом Кернига, при котором пострадавший не может полностью разогнуть ногу.
Клиника ДАП включает ряд вегетативных проявлений:
- повышенное кровяное давление;
- потливость;
- слюнотечение.
У многих получивших травму согнутые в локтях руки прижаты к телу, кисти свободно свисают, подобно лапкам грызунов или кенгуру. Сухожильные рефлексы сначала повышаются, но после снижаются или исчезают вовсе.
Нарушается тонус мышц, что может проявиться в виде приступов мышечного гипертонуса в руках или ногах. Конечности плотно прижимаются к телу или хаотично сгибаются-разгибаются.
3 степени тяжести
|
Степень тяжести |
Длительность комы |
Интенсивность симптоматики |
|
1 – легкая |
От 6 часов до суток |
Симптомы слабовыраженные |
|
2 – средняя (умеренная) |
Больше суток |
Грубые стволовые признаки отсутствуют |
|
3 – тяжелая |
Продолжительная кома, которая может длиться годами |
Тяжелый неврологический дефицит, высокий риск летальности |
Лечится ли диффузное состояние
Все зависит от степени тяжести заболевания. В последние годы появилось мнение, что у детей и молодых людей аксоны способны саморегенерироваться. Это подтверждается частичным восстановлением неврологических функций у пациентов с диффузным повреждением аксонов мозга.
Диффузное аксональное повреждение головного мозга в детском и молодом возрасте более распространено, по сравнению со взрослыми. Учитывая анатомо-физиологические особенности детского организма, процессы, протекающие после черепно-мозговых травм, имеют значительные отличия.
Пример из практики:
Женщина 28 лет госпитализирована в отделение травматологии в бессознательном состоянии крайней степени тяжести. Она пострадала в автомобильной аварии, ее машина несколько раз переворачивалась. Диагноз при поступлении – тяжелый ушиб головного мозга. Назначено лечение, пациентка пришла в сознание через 27 часов после ДТП. Данные МРТ соответствуют классическим признакам диффузного аксонального повреждения мозга средней степени тяжести.
У детей чаще восстанавливается сознание, хотя бы на время, после сравнительно легких ЧМТ, и более вероятно быстрое улучшение состояния. Помимо этого, прогноз у них благоприятнее, чем можно предположить с учетом исходного неврологического дефицита.
Тем не менее, риск тяжелой инвалидизации тем выше, чем дольше продолжается кома, и чем выраженнее неврологические симптомы. Если, конечно, пострадавший выживает.
Диффузное аксональное поражение мозга с комой продолжительностью не больше недели прогностически довольно благоприятно. Восстановление идет хорошо, инвалидизация умеренная.
Если кома длится больше 8 дней, высок риск тяжелой инвалидности либо перехода в вегетативное состояние.
Последствия
У людей, получивших ЧМТ по типу диффузии, возможно 2 исхода:
- Выход из коматозного состояния.
- Переход в вегетативное состояние.
Выход из комы
Если пациент открывает глаза, способен фиксировать взгляд на лицах и предметах, а также следить за ними глазами, это означает выход из комы. Спровоцировать его может все, что угодно – звук, яркий свет, боль.
Внимание! Сознание постепенно проясняется, к пациенту возвращается способность выполнять простейшие действия и разговаривать. Параллельно с этим регрессирует неврологическая симптоматика.
Переход в вегетативное состояние
Продолжительная кома чаще всего ведет к переходу в вегетативное состояние, которое может быть стойким или иметь эпизодический характер. В последнем случае его длительность составляет от 1 дня до нескольких месяцев.
В медицине вегетативное состояние определяется, прежде всего, по открыванию глаз с отсутствием фокусирования взгляда. Долговременное пребывание в вегетативном состоянии сопровождается такими симптомами, как:
- учащение сердечного ритма;
- повышенная температура тела;
- частое поверхностное дыхание – тахипноэ;
- образование пролежней;
- полиорганная недостаточность;
- застойная пневмония, сепсис, почечные инфекции с развитием пиелонефрита.
В результате диффузного аксонального повреждения в мозге возникают необратимые изменения, влекущие за собой остаточные явления, среди которых могут быть:
- парезы и параличи;
- частичная потеря памяти;
- психические отклонения;
- речевые дефекты;
- симптомы бульбарного синдрома – нарушение глотания, кашель и поперхивание во время приема пищи, слабость голоса, гнусавость;
- двигательные расстройства (тики, мышечные спазмы, тремор конечностей и т. д.).
При выходе из вегетативного состояния серьезно страдает психика: развивается слабоумие, лабильность настроения, апатичность, появляются трудности с запоминанием текущих событий.
Продолжительное вегетативное состояние заканчивается летально.
![]()
Агишев Дамир Адгемович
врач – невролог
Запишитесь на онлайн-консультацию, если ваш родственник или близкий человек находится в коме или вегетативном состоянии. Наши врачи на дистанционной консультации расскажут, как вести себя с пострадавшим, и что можно для него сделать. Специалисты на связи круглосуточно.
Online консультация
Частые вопросы
Лечится ли травма головного мозга с повреждением аксонов хирургически?
+
Оперативное вмешательство имеет строгие показания и проводится при обнаружении гематом и размозжения, компрессионных переломов черепных костей.
Какие препараты назначают при диффузных изменениях сосудов?
+
Такие препараты входят в стандартную схему терапии и включают Винпоцетин, Циннаризин.
Какие исследования проводятся для подтверждения диагноза ДАП?
+
Наиболее информативными являются компьютерная и магнитно-резонансная томография.
Как лечат пациентов в коме?
+
Пострадавших обязательно подключают к аппарату искусственной вентиляции легких, они получают питание парентерально. Интенсивное лечение направлено на поддержание работы внутренних органов, ликвидацию отека мозга, предотвращение инфекций. Избыточная двигательная активность является показанием к применению наркоза.
Заключение эксперта
При обследовании пострадавших важно учитывать характер травмы (резкое ускорение или торможение головы, приводящее к натяжению и разрыву аксонов), продолжительность комы и имеющиеся симптомы, а также данные МРТ. От точности диагностики зависит правильность назначения и исход лечения.