 Верхние нейроны головного мозга, отвечающие за движение, расположены в полушарной коре, спускаются и контактируют в бульбарном отделе своими отростками с клетками спинного мозга.
Верхние нейроны головного мозга, отвечающие за движение, расположены в полушарной коре, спускаются и контактируют в бульбарном отделе своими отростками с клетками спинного мозга.
Мотонейроны спинного мозга также распространены в поясничном, грудном и шейном отделах (исходя из типа мышц, куда направлены сигналы, регулирующие их сокращения). Двигательный нейрон проводит нервный импульс от спинного мозга к коре головного.
Болезнь двигательного нейрона (БДН) является нейродегенеративной патологией с нарастающим характером, в результате чего повреждаются мотонейроны спинного и головного мозга. В результате постепенного отмирания нервных волокон нарастает мышечная слабость с усилением симптоматики и летальным исходом.
Из-за повреждения моторных нейронов пропадают функции спинного мозга и позвоночника, постепенно наступает паралич, теряется речь, затрудняется глотание.
Уровень заболеваемости составляет 2-5 человека к 100 тысячам людей ежегодно. Заболевание проявляется в возрасте 50-70 лет, иногда у пациентов моложе 40 лет. В среднем пациенты живут 1-4 года после постановки диагноза.
Поражаемые области
С постепенным развитием заболевания двигательных нейронов у пациентов проявляется нарастание проблем с дыханием, глотанием, движением и общением с другими людьми. Могут проявляться следующие затруднения:
- Кисти, руки. Даже при выполнении ежедневных дел (мытья посуды, поворачивание крана, нажатие клавиш) будут проявляться все большие трудности с усилением слабости в ладонях и руках.
- Ноги. Из-за ослабления ног пациенту будет все труднее передвигаться.
- Эмоциональное состояние. Иногда могут поражаться зоны, которые отвечают за эмоциональные реакции, что проявляется непроизвольным смехом или плачем физиологического характера.
- Плечи, шея. Через определенное время пациенту становится трудно дышать из-за слабеющих мышц шеи.
- Дыхание. В основном заболевание распространяется на дыхательные мышцы, затрудняя дыхание.
- Глотание, речевые функции. Могут быть затруднения с приемом пищи, питьем и разговором.
У некоторых пациентов БДН вызывает нарушения памяти, сниженную концентрацию внимания, сложности с выбором слов и обучаемостью. То есть происходит незаметное и скрытое изменение психических функций.
При БДН остается не нарушенным интеллект, пациенты осознают все происходящее. У многих людей не поражается зрение, вкус, осязание и слух, остаются работоспособными функции мочевого пузыря, кишечника, половых органов (вплоть до поздних стадий) и сердечной мышцы.

Патогенез и патоморфология болезни
Болезнь мотонейрона считается патологией мультифакторного типа, развитие которой происходит от воздействия генетической наследственности с окружающими факторами.
В 10% ситуаций болезнь связана с семейными формами, где 25% — характеризуются мутированием гена СОД-1. Иногда спорадические виды патологии связаны с изменениями других генов (нейрофиламентов) в результате нарушений функций или структуры, при действии которых мотонейроны начинают дегенерировать. Механизм запускается при действии провокаторов (возраста, пола, тяжелых металлов и пр.). Маловероятной является взаимосвязь появления заболевания от инфекционных агентов.
Клинически нейроны дегенерируют при разрушении от 80% клеточных структур, что затрудняет своевременную терапию патологии и нуждается в диагностических исследованиях во время доклинического этапа.
Возле лобных долей, в 3, 5 слоях центральных извилин, двигательных ядрах 5, 7, 9 и 11 нервов черепа в стволе мозга определяется дегенерация нейронов и изменение кортикоспинальных путей. На различных этапах изменения мотонейронов определяются эозинофильные, базофильные включения и тельца Буниной.
Аксональные сфероиды образуются а проксимальных отделах аксонов. На основании полученных данных, отмечаются нарушения перемещения и деградации белков. В мышечных структурах определяется денервационная атрофия.
Что является причиной гибели двигательных нейронов?
По мнению специалистов, основными причинами БДН является наследственность и средовые факторы, повышающие риск патологии.
 Примерно у 5-10% пациентов с болезнью моторного нейрона была выявлена семейная наследственная форма. В основном БДН была диагностирована (90-95% случаев) при спорадической форме, которая появляется по неизвестным причинам.
Примерно у 5-10% пациентов с болезнью моторного нейрона была выявлена семейная наследственная форма. В основном БДН была диагностирована (90-95% случаев) при спорадической форме, которая появляется по неизвестным причинам.
По эпидемиологическим исследованиям была выявлена возможная связь действия шоковых, механических травм, высоких спортивных нагрузок, вредных веществ на развитие заболевания.
Сейчас проводятся обследования механизмов повреждения внутриклеточных процессов, из-за которых погибают двигательные нейроны с окружающими клетками.
К возможным причинам относят забастовку и сбой работы «редакторов» в обработке РНК, изменения химических коммуникативных связей спинного мозга, нарушения транспортировки продуктов метаболизма, питательных веществ, скопления агрегатов белка в нейронах с нарушением нормальных функций и склеиванием, появление свободных радикалов кислорода, нарушение энергетического фона клеток и недостаток питательных веществ для мотонейронов. Также гибель двигательных нейронов может возникать из-за окружения глиальными клетками, которые могут быть токсичными.
Основные и редкие виды БДН
Заболевание двигательных нейронов имеет несколько основных форм:
- БАС (боковой амиотрофический склероз) развивается у 80% людей с болезнью двигательного нейрона, вызывает в основном слабость, судороги и истощение в мышцах, скованность верхних и нижних конечностей;
- ПБП (бульбарный паралич прогрессирующего типа) поражает около 10-25% людей, распространяется на нейроны спинного и головного мозга, проявляется затруднениями в жевании, глотании, неразборчивости речи;
- мышечная прогрессирующая атрофия (8%) – редкий тип БДН, распространяется на двигательные нейроны в нижних конечностях, развивается слабость, атрофия мышц, фасцикуляции и потеря веса;
- ПЛС (латеральный первичный склероз) (2%) поражает преимущественно двигательные нейроны в верхних конечностях, развивая спастику мышц, неустойчивую ходьбу, затрудненную речь.

Симптоматика и клиника БДН в зависимости от формы
На моторные нейроны влияет целая группа болезней. В состав нейронов входят две группы клеток, необходимые для произвольных движений.
В первую группу входят двигательные нейроны, расположенные в прецентральной мозговой извилине, а во вторую – нейроны, расположенные в стволе спинного и головного мозга.
В зависимости от локализации погибшего двигательного нейрона будут зависеть форма болезни и ее симптомы.
При спастической параплегии поражается двигательный нейрон первой группы, при спинальной мышечной атрофии, детском параличе – вторая группа, а при боковом амиотрофическом склерозе обе группы нейронов.
Формы БДН и их проявления:
-
Спастическая параплегия определяется повышением спастического тонуса ног с нарушением походки. Это вызывается
 генетическими мутациями.
генетическими мутациями. - Для спинальной мышечной атрофии характерна слабость и уменьшение мышц, отсутствие их активации или иннервирования нервными волокнами. В зависимости от времени появления первой симптоматики и характеру распределения слабости мышц, осуществляется классификация спинальной мышечной атрофии.
- Более распространенной патологией опорно-двигательной системы является БАС, частота развития которого повышается с возрастом. В результате прогрессирует омертвление нервных клеток, нарастает слабость и мышечная атрофия.
- Детский церебральный паралич, полиомиелит является вирусной болезнью нижних нейронов.
 генетическими мутациями.
генетическими мутациями.Дифференциальная диагностика
 Диагноз БДН подтверждается элеткронейромиографией, которая выявляет генерализованный денервационный процесс. Для выявления спонтанности, продолжительности двигательных единиц, фасцикуляций, фибрилляций, выполняется трехуровневая игольчатая ЭМГ.
Диагноз БДН подтверждается элеткронейромиографией, которая выявляет генерализованный денервационный процесс. Для выявления спонтанности, продолжительности двигательных единиц, фасцикуляций, фибрилляций, выполняется трехуровневая игольчатая ЭМГ.
Стимуляционная электронейромиография позволяет выявлять замедление двигательных волокон, пирамидную недостаточность.
Транскраниальная магнитная стимуляция выявляет уменьшение возбудимости в центральных мотонейронах. МРТ позволяет исключать остальные патологии с похожей клиникой (компрессию грыжи или опухолью).
Возможности лечения
Болезнь двигательного нейрона неизлечима, а целью терапии является замедление развития процесса, сокращение выраженности симптоматики, так как не существует методов для полного излечения. Доказана эффективность Рилутека – пресинаптического ингибитора.
Как и при БАС, могут применяться антиоксидантные средства. Также специалисты назначают креатин, карнитин, витамины. Другим лекарственным средством является Рилузол, но средство официально недоступно для пациентов в РФ. Как и Рилутек, препарат снижает объем глутамата, освобождающегося при передаче нервных импульсов.
К полиативной терапии относится устранение болей от мышечных спазмов, для чего назначается Карбамазепин в дозе около 100-200 мг несколько раз в сутки. При увеличенном тонусе мышц нужно принимать миорелаксанты (Сирдалуд, Баклофен).
Дыхательная поддержка – это основной фактор, влияющий на качество жизни и прогноз. С прогрессированием патологии наблюдается ослабление и атрофия мышц диафрагмы и других мышечных групп, требуется неинвазивное вентилирование легких.
Это нормализует сон, снижает утомляемость и одышку. Используется аппарат со специальной маской.
Если нарушены функции глотания, то кормление проводится назогастральным зондом, установкой гастростомы. Назначается специальное питание, восполняющее недостаток калорий.
При заболевании БДН трудно говорить про прогноз. В редких случаях люди живут десятки лет. Человек может умереть через несколько месяцев. Но в среднем длительность жизни равна 2-5 года от начала выявления болезни. Не существует специфических профилактических мер.
БАС — двигательные нейроны отмирают полностью
Во время БАС сочетается равномерно выраженная симптоматика центрального и периферического паралича, преобладание одних симптомов над другими (сегментарно-ядерный или пирамидный варианты). На поздних этапах болезни преобладают показатели периферического паралича.
Во время БАС начинается повреждение мотонейронов в передних рогах спинового мозга. С прогрессированием патологии проявляются бульбарные нарушения в основном у мужчин.
Выделяется грудной, шейный, диффузный и поясничный дебют. Классическим вариантом является шейный, первая симптоматика которого характеризуется слабостью и атрофией кисти, нарушенными мелкими движениями, наличием фасцикулярных подергиваний пораженных мышц.

Стивен Хокинг — самый известный пациент с БАС
В дальнейшем появляется слабость и атрофия мышц плечевого пояса, пациентам сложно одеваться, поднимать руки выше горизонтальной плоскости. Сначала поражается одна сторона тела, затем симптомы проявляются на другой руке с развитием верхнего смешанного парапареза.
Развивается слабость и скованность нижних конечностей, больным сложно ходить на большие расстояния, перемещаться по лестнице.
Повреждаются разгибательные мышцы с проявлением смешанного нижнего парапареза. На поздних стадиях проявляются псевдобульбарный и бульбарный синдромы, появление дисфагии.
Первой симптоматикой грудного дебюта будут фасцикуляции, атрофия мышц в животе, спине и слабость. Пациентам становится сложно стоять и нагибаться. На следующих этапах появляется смешанный односторонний гемипарез с вовлечением мускулатуры противоположной части тела. На начальном этапе выпадают брюшные рефлексы. В результате дыхательных нарушений наступает летальный исход.
Диффузный дебют проявляется признаками нарушения периферических мотонейронов с резко выраженным утомлением, дыхательными нарушениями. Снижается масса тела до появления дисфагии, в результате дыхательной недостаточности наступает смерть.
Обычно поражение жевательных и мимических мышц наступает позже, пациенту сложно высовывать язык, вытягивать в трубочку губы, надувать щеки.
К осложнениям БАС относят параличи, парезы шейных мышц и конечностей, нарушенное глотание, дыхательную недостаточность, контрактуры конечностей, аспирационную пневмонию, уросепсис, депрессию, болезненные спазмы мышц, кахексию. С прогрессированием двигательных нарушений пациент умирает.
Проведение диагностики и лечение
Диагностика БАС основывается на детальном анализе клиники. Проводится электронейромиография (ЭМГ обследование) для подтверждения нарушений мотонейрона.
Не существует эффективного лечения патологии. Можно отсрочить летальный исход на несколько месяцев приемом Рилузола в дозировке 50 мг 2 раза в день. Основой терапии является лечебная гимнастика, физическая активность, диета и пр.
Болезненные спазмы мышц устраняются Дифенином, Хинина сульфат, Тегретолом, Финлепсином, витамином Е400 мг, Изоптином или  препаратами магния. При слюнотечении назначается Атропин, Бускопн, при спастичности – Сирдалуд, Баклосан, Мемантин и пр.
препаратами магния. При слюнотечении назначается Атропин, Бускопн, при спастичности – Сирдалуд, Баклосан, Мемантин и пр.
Болевые симптомы устраняются анальгетиками. Временное улучшение может быть от антихолинэстеразных средств. Также специалисты назначают антидепрессанты (Амитриптилин, Серталин).
Пациент должен поддерживать физическую активность, при прогрессировании патологии использовать специальные приспособления. В результате дисфагии может потребоваться питание через зонд или гастростомия. В последнее время проводятся исследования лечения патологии стволовыми клетками.
БАС считается фатальной патологией, средняя продолжительность жизни при которой составляет 3-5 лет. Только около 10% пациентов живут в течение 10 лет. К негативным прогностическим показателям относят бульбарные нарушения, пожилой возраст.
| Боковой амиотрофический склероз | |
|---|---|
 |
|
| МКБ-10 | G12.2 |
| МКБ-10-КМ | G12.21 и G12.2 |
| МКБ-9 | 335.20 |
| МКБ-9-КМ | 335.20 |
| OMIM | 105400 |
| DiseasesDB | 29148 |
| MedlinePlus | 000688 |
| eMedicine | neuro/14 |
| MeSH | D000690 |
Боково́й (латера́льный) амиотрофи́ческий склеро́з (БАС; также известен как боле́знь мото́рных нейро́нов, мотонейро́нная боле́знь, боле́знь Шарко́, в англоязычных странах — болезнь Лу Ге́рига [англ. Lou Gehrig’s disease]) — прогрессирующее, неизлечимое дегенеративное заболевание центральной нервной системы, при котором происходит поражение как верхних (моторная кора головного мозга), так и нижних (передние рога спинного мозга и ядра черепных нервов) двигательных нейронов, что приводит к параличам и последующей атрофии мышц.
Характеризуется прогрессирующим поражением двигательных нейронов, сопровождаемым параличом (парезом) конечностей и атрофией мышц. Смерть наступает от инфекций дыхательных путей или отказа дыхательной мускулатуры. Боковой амиотрофический склероз следует отличать от синдрома БАС, который может сопровождать такие заболевания, как клещевой энцефалит.
Болезнь впервые описана в 1869 году Жан-Мартеном Шарко.
На международном уровне показатели заболеваемости боковым амиотрофическим склерозом (amyotrophic lateral sclerosis, ALS) или заболеванием двигательных нейронов (motor neurone disease, MND) по всему миру оцениваются в диапазоне от 0,86 до 2,5 на 100 тысяч человек в год, то есть БАС является редким заболеванием.
Содержание
- 1 Этиология
- 2 Факторы риска
- 3 Течение болезни
- 4 Симптомы
- 5 Диагностика
-
6 Лечение
- 6.1 Замедление прогрессирования
- 6.2 В России
- 7 Акции в поддержку
- 8 См. также
- 9 Примечания
- 10 Ссылки
Этиология
Точная этиология БАС неизвестна. Примерно в 5% случаев встречаются семейные (наследственные) формы заболевания. 20% семейных случаев БАС связаны с мутациями гена супероксиддисмутазы-1, расположенного в 21-й хромосоме. Как полагают, этот дефект наследуется аутосомно-доминантно.
В патогенезе заболевания ключевую роль играет повышенная активность глутаматергической системы, при этом избыток глутаминовой кислоты вызывает перевозбуждение и гибель нейронов (т. н. эксайтотоксичность). Сохранившиеся мотонейроны могут спонтанно деполяризироваться, что клинически выявляется фасцикуляциями.
Учёные из Университета Джонса Хопкинса в Балтиморе установили молекулярно-генетический механизм, лежащий в основе возникновения данного заболевания. Он связан с появлением в клетках большого количества четырёхспиральной ДНК и РНК в гене C9orf72, что приводит к нарушению процесса транскрипции, а, следовательно, и синтеза белка. Однако, вопрос о том, как именно эти изменения ведут к деградации мотонейронов, остаётся открытым.
Также важное значение в патофизиологии имеет TDP-43, который был определен как основной компонент убиквитинированных цитоплазматических белковых агрегатов у всех пациентов со спорадическим БАС, но расположенный вне ядра (в нормальных нейронах он находится в ядре). Несмотря на то, что вопрос того, являются ли данные агрегаты причиной нейродеградации при БАС, остается открытым, мутации в TARDBP были обнаружены всего лишь в 3 % случаях наследственной формы склероза и у 1,5 % пациентов со спорадическим БАС, позволяя предположить, что агрегаты TDP-43 играют ключевую роль в инициации БАС. Помимо мутаций в гене TARDBP ионы цинка также могут вызывать агрегацию TDP-43.
Обнаружение мутаций гена FUS (Fusion in Sarcoma — ген «слияния в саркоме») в 16-й хромосоме, которые связаны с наследственными формами БАС, поддерживает данную теорию. Агрегаты FUS не были явно определены у пациентов с патологическими изменениями в TDP-43 или SOD1, что указывает на новый путь возникновения болезни.
Факторы риска
На БАС приходится примерно 3% всех органических поражений нервной системы. Болезнь обычно развивается начиная с возраста 30—50 лет.
Суммарный риск получить БАС в течение жизни составляет 1:400 для женщин и 1:350 для мужчин.
5—10 % заболевших — носители наследственной формы БАС; на тихоокеанском острове Гуам выявлена особая, эндемичная форма заболевания. Абсолютное большинство случаев (90—95 %) не связаны с наследственностью и не могут быть положительно объяснены какими-либо внешними факторами (перенесёнными заболеваниями, травмами, экологической ситуацией и т. п.).
Несколько научных исследований нашли статистические корреляции между БАС и некоторыми сельскохозяйственными пестицидами.
Течение болезни
Ранние симптомы болезни: подёргивания, судороги, онемение мышц, слабость в конечностях, затруднение речи — также свойственны многим более распространённым заболеваниям, поэтому диагностика БАС затруднена — до тех пор, пока болезнь не развивается до стадии мышечной атрофии.
В редких случаях возможно наличие продромальной фазы, до 1 года, во время которой будут наблюдаться изолированные фасцикуляции и/или судороги.
В зависимости от того, какие части тела поражены в первую очередь, различают
- БАС конечностей (до трёх четвертей больных) начинается, как правило, с поражения одной или обеих ног. Больные чувствуют неловкость при ходьбе, негибкость в голеностопе, спотыкаются. Реже встречаются поражения верхних конечностей, при этом затруднено выполнение обычных действий, требующих гибкости пальцев или усилия кисти.
- Бульбарный БАС проявляется в затруднении речи (больной говорит «в нос», гнусавит, плохо управляет громкостью речи, в дальнейшем испытывает трудности с глотанием).
Во всех случаях мышечная слабость постепенно охватывает всё больше частей тела (больные бульбарной формой БАС могут не доживать до полного пареза конечностей). Симптомы БАС включают признаки поражения как нижних, так и верхних двигательных нервов:
- поражение верхних двигательных нейронов: гипертонус мышц, гиперрефлексия, аномальный рефлекс Бабинского.
- поражение нижних двигательных нейронов: слабость и атрофию мышц, судороги, непроизвольные фасцикуляции (подёргивания) мышц.
Рано или поздно больной теряет способность самостоятельно передвигаться. Болезнь не влияет на умственные способности, но приводит к тяжёлому состоянию в ожидании неминуемой смерти. На поздних этапах болезни поражается дыхательная мускулатура, больные испытывают перебои в дыхании, в конечном итоге их жизнь может поддерживаться только искусственной вентиляцией лёгких и искусственным питанием. Обычно от выявления первых признаков БАС до смерти проходит от трёх до пяти лет. Однако широко известный физик-теоретик Стивен Хокинг (1942-2018) и гитарист Джейсон Беккер (род. 1969) — единственные известные больные с однозначно диагностированным БАС, у которых состояние со временем стабилизировалось.
Симптомы
- слабость;
- мышечные спазмы;
- нарушения речи и глотания;
- нарушение равновесия;
- спастика;
- повышение глубоких рефлексов или расширение рефлексогенной зоны;
- патологические рефлексы;
- атрофия;
- зависание стопы;
- респираторные расстройства;
- приступы непроизвольного смеха или плача;
- депрессия.
Диагностика

Существует множество заболеваний, вызывающих те же симптомы, что и ранние стадии БАС. Диагностика заболевания возможна только методом исключения более распространённых заболеваний. Оба ключевых признака БАС (поражения и верхних, и нижних двигательных нейронов) проявляются на достаточно развитых стадиях болезни.
Международной федерацией неврологии (англ. World Federation of Neurology) разработаны Эль-Эскориальские критерии для постановки диагноза БАС. Для этого необходимо наличие:
- признаков поражения центрального мотонейрона по клиническим данным
- признаков поражения периферического мотонейрона по клиническим, электрофизиологическим и патоморфологическим данным
- прогрессирующего распространения симптомов в пределах одной или нескольких областей иннервации, что выявляют при наблюдении за больным
При этом должны быть исключены другие причины данных симптомов.
Для электрофизиологического обследования используется электромиография, которая полезна в исследовании проводимости нервов и определении наличия признаков поражения периферического мотонейрона (потенциалы фибрилляций, потенциалы фасцикуляций, положительные острые волны и др).
Также важно дифференцировать фасцикуляции при БАС от фасцикуляций при синдроме доброкачественных фасцикуляций (англ. BFS), который зачастую диагностируется при наличии фасцикуляций и одновременном отсутствии объективной слабости и изменений на ЭМГ, и имеет чаще всего психологическую причину.
Второстепенными методами диагностики являются:
- МРТ головного и спинного мозга
- биохимический анализ крови (КФК, креатинин, общий белок, АЛС, АСТ, ЛДГ)
- клинический анализ крови
- исследование ликвора (белок, клеточный состав)
- серологические анализы (антитела к боррелиям, к ВИЧ)
- транскраниальная магнитная стимуляция (ТМС)
- биопсия мышцы или нерва
Возможные изменения при обследовании второстепенными методами диагностики:
- повышение КФК в 2-3 раза (у 50 % больных)
- незначительное повышение АЛТ, АСТ, ЛДГ
- выявление отмирания пирамидных путей на МРТ
- признаки атрофии и денервации при гистологическом исследовании
Лечение
Больным БАС требуется поддерживающая терапия для облегчения симптомов.
Постепенно у больных начинает ослабляться дыхательная мускулатура, развивается дыхательная недостаточность и становится необходимым применение оборудования для облегчения дыхания во время сна (IPPV или BIPAP). Затем, после полного отказа дыхательной мускулатуры, требуется круглосуточное использование аппарата искусственной вентиляции лёгких.
Ведутся исследования по методу лечения, использующего блокировку генов, вызывающих это заболевание.
Замедление прогрессирования
Рилузол (рилутек) — единственный препарат, достоверно замедляющий прогрессирование БАС. Доступен с 1995 года. Он ингибирует высвобождение глутамата, тем самым уменьшая повреждение двигательных нейронов. Продлевает жизнь больных в среднем на месяц, немного отдаляет момент, когда больному потребуется искусственная вентиляция легких.
HAL-терапия, новый метод роботизированного лечения, официально допущен к применению в реабилитации БАС в Европе и Японии.
Также проводятся клинические исследования по применению препаратов Radicava и Masitinib.
В России
В Москве действуют:
- Благотворительный фонд помощи людям с БАС и другими нейромышечными заболеваниями «Живи сейчас»
- Благотворительный фонд помощи больным БАС Г. Н. Левицкого. http://www.alsportal.ru
- служба помощи больным БАС при АНО «Больница Святителя Алексия».
В то же время в России многим больным БАС не оказывается надлежащая медицинская помощь. Например, до 2011 года БАС даже не был включен в список редких заболеваний, а единственный препарат, замедляющий течение болезни, Рилузол, не зарегистрирован.
Акции в поддержку
Летом 2014 года проходила популярная вирусная акция по повышению осведомлённости о заболевании и сбор средств, получившая название Ice Bucket Challenge или ALS Ice Bucket Challenge. Летом 2018 года акция прошла повторно для сбора средств на построение клиники по борьбе с заболеванием в Южной Корее.
См. также
- Супероксиддисмутаза-1 — фермент, связанный с частью случаев заболевания.
- Спинальная мышечная атрофия
Болезнь двигательного нейрона по МКБ-10 относится к классу болезней с системной атрофией элементов центральной нервной системы. Болезнь двигательного нерва (БДН) также называют боковой амиотрофический склероз (БАС) или болезнь моторных нейронов. Это заболевание характеризуется поражением верхних нейронов головного мозга с последующим нарушением их связи со спинным мозгом. В результате заболевания развивается паралич всего тела.
На сегодняшний день БДН относится к неизлечимым заболеваниям. Все терапевтические средства направлены на замедление прогрессирования патологического процесса и улучшение качества жизни пациента. В Юсуповской больнице высококвалифицированные неврологи выполняют поддерживающую терапию пациентов с БДН, которая способствует облегчению их состояния.

Причины
Исходя из того, что болезнь двигательного нейрона по МКБ-10 относят к системным атрофиям с поражением преимущественно центральной нервной системы, основными ее характеристиками являются дегенеративные процессы в центральных извилинах лобных долей.
Разрушительный процесс распространяется на ядра двигательных нейронов ствола головного мозга с изменением в кортикоспинальных путях. В результате происходит медленно прогрессирующая мышечная атрофия.
Причинами развития БДН являются наследственность и факторы окружающей среды. В семейных случаях развития патологии отмечаются мутации отдельных генов, кодирующих ДНК-связывающие белки. Наследственная форма патологии встречается в 5-10% случаев. В остальных случаях причина патологии остается неизвестной.
Провоцирующими факторами могут быть:
- шоковые и механические травмы головного и спинного мозга;
- чрезмерные физические нагрузки;
- воздействие вредных веществ.
На данный момент болезнь двигательного нерва продолжает изучаться. Сейчас разрабатываются теории о связи патологии с нарушением работы химических структур РНК, сбоем транспортировки продуктов метаболизма в клетках, нарушением энергетического обмена.
Исследования показали, что инфекционные заболевания, такие как полиомиелит, не вызывают болезнь двигательного нерва, хотя и характеризуются прогрессирующей мышечной атрофией. Патологии хоть и имеют примерно одинаковый конечный результат, но механизмы их действия разные.
Болезнь двигательного нейрона: виды
Болезнь двигательного нейрона имеет код МКБ-10 G12: Спинальная мышечная атрофия и родственные синдромы. Она относится к группе неврологических заболеваний, при которых происходит поражение двигательных нейронов. Синонимы: болезнь Лу Герига, болезнь Шарко. Во многих станах для обозначения болезни двигательного нейрона принято употреблять термин «Боковой амиотрофический склероз» (БАС), как наиболее распространенной патологии в этой группе. Сюда также относят:
- наследственную спастическую параплегию;
-
первичный боковой склероз;
-
прогрессирующую мышечную атрофию;
-
болезнь двигательного нейрона, бульбарная форма;
-
псевдобульбарный паралич;
-
первичный латеральный склероз.
Болезнь характеризуется дегенерацией двигательных нейронов в коре головного мозга, стволе головного мозга, кортикоспинальных путях и спинном мозге. В результате происходит прогрессирующий мышечный паралич.
Заболевание относится к редким. Его распространенность составляет примерно 2-3 человека на 100 тысяч в год. Наиболее часто болезнь возникает у людей в возрасте 60-70 лет, хотя не исключено развитие патологии и у людей младше 40 лет.
Симптомы
Прогрессирующая мышечная атрофия вызывается дистрофическими процессами в моторных нейронах. Клинические проявления будут зависеть от того, в какой группе клеток произошло нарушение. Например, болезнь двигательного нейрона с боковым амиотрофическим склерозом возникает в результате поражения нейронов в прецентральной мозговой извилине и стволе спинного мозга. Спастическая параплегия развивается при нарушениях в прецентральной мозговой извилине. Прогрессирующий бульбарный паралич происходит на фоне изменений в работе бульбарной группы каудальных нервов с поражением их ядер и корешков.
С прогрессированием патологии у пациента возникают двигательные нарушения, проблемы с глотанием, дыханием, ухудшаются коммуникативные навыки. Болезнь имеет следующую характерную симптоматику:
- ухудшается работа рук, мелкая моторика. Пациенту трудно выполнять самые простые задачи: открыть ручку двери или кран, взять предмет, нажать на кнопку;
- нарастает слабость в ногах, что приводит к трудностям в передвижении;
- ослабевают мышцы шеи, вследствие чего пациенту становится трудно дышать, глотать пищу и воду;
- болезнь часто затрагивает мышцы, которые участвуют в процессе дыхания;
- при поражении зон головного мозга, ответственных за эмоции, пациент может не контролировать свое поведение, периодически беспричинно смеясь или плача.
С прогрессированием патологии ухудшается память, снижается концентрация внимания, затрудняется общение с другими людьми. На поздних стадиях пациент становится полностью обездвижен и ему требуется постоянных уход. При этом болезнь не затрагивает интеллект, больной продолжает ощущать вкус и запах, слух и зрение не страдают. Функции сердечной мышцы, кишечника, мочевого пузыря, половых органов остаются без изменений.

Боковой амиотрофический склероз (БАС)
Впервые заболевание описано французом Жаном-Мартеном Шарко в 1869 году. Это самая распространенная форма заболевания (около 80-85% всех случаев БДН), когда в патологический процесс вовлечены двигательные нейроны и головного, и спинного мозга.
Болезнь характеризуется неуклонно прогрессирующей мышечной слабостью и атрофиями мышц. В зависимости от уровня локализации первичного поражения двигательных нейронов различают бульбарную форму заболевания, когда первично повреждаются мотонейроны в стволе головного мозга, шейно-грудную форму – при локализации первичного очага поражения на уровне шейного утолщения и пояснично-крестцовую форму заболевания. Наиболее благоприятным вариантом течения считается пояснично-крестцовая форма БАС. Средняя продолжительность жизни таких больных может достигать 7-8 лет. В некоторых случаях длительность заболевания достигает 10 или даже 15 лет. Болезнь развивается, как правило, на 5-6 десятке жизни и в 2 раза чаще встречается у мужчин.
Дебютирует болезнь в большинстве случаев с появления слабости и похудания в мышцах рук или ног, часто сопровождается мышечными подергиваниями (фасцикуляциями), однако следует помнить, что наличие изолированных мышечных подергиваний без появления слабости или гипотрофий не всегда является признаком БАС!
Среди известных людей БАС страдали композитор Дмитрий Шостакович, китайский политический деятель Мао Цзэдун, певец Владимир Мигуля, американский астрофизик Стивен Хокинг, длительность заболевания у которого превышает 50 лет.
Прогрессирующий бульбарный паралич (ПБП)
Основное отличие ПБП от других видов болезни двигательного нейрона — быстро нарастающие нарушения речи и глотания. Локализация первичного процесса — ствол головного мозга. Продолжительность жизни колеблется между 6 месяцами и 3 годами с начала появления симптомов.
Первичный латеральный склероз (ПЛС)
Редкая форма БДН, затрагивающая исключительно двигательные нейроны головного мозга, что выражается нарастанием спастичности в конечностях. Пациент практически не может самостоятельно согнуть руку или ногу вследствие высокого тонуса. Отличительными особенностями данного вида БДН является отсутствие гипотрофий, фасцикуляций и речевых нарушений. Длительность болезни, как правило, составляет более 10 лет.
Прогрессирующая мышечная атрофия (ПМА), синдром свисающих рук
Это редкий вид БДН, при котором в основном повреждаются двигательные нейроны спинного мозга. Заболевание в большинстве случаев начинается со слабости или неловкости в руках. Большинство людей живут с этим видом БДН более 5 лет.
Здесь приведены наиболее часто встречающиеся симптомы и характеристики разных видов БДН. Однако необходимо помнить, что при одном и том же виде болезни двигательного нейрона симптомы у разных людей могут проявляться по-разному, прогноз также может отличаться.
При появлении таких симптомов, как постепенно нарастающие речевые нарушения, поперхивание при глотании, слабость в руках и ногах, мышечные подергивания, следует, как можно раньше обратиться за медицинской помощью к высококвалифицированным неврологам Юсуповской больницы, которые помогут определить причины и назначить адекватное лечение.
Необходимо понимать, что диагностика БАС весьма затруднительна, так как это достаточно редкое заболевание, и зачастую неврологи в своей практике встречают единичные случаи данного заболевания. Отсутствие определенных специфических симптомов заболевания также затрудняет диагностику БАС. Известно, что диагноз устанавливается в среднем через 1-1,5 года от момента появления первых симптомов, и время для лечения уже упущено.
Диагностика
Существуют несколько методов и показателей диагностики, которые указывают на страдание двигательных нейронов и мышц.
Анализ крови
Основным показателем, который может указать на страдание двигательных нейронов и мышц – это уровень креатинфосфокиназы. Уровень фермента увеличивается при распаде мышечной ткани, и его уровень может быть повышен у людей, страдающих БАС. Однако данный показатель не является специфическим признаком патологии, поскольку может быть индикатором и других заболеваний, связанных с поражением мышц (инфаркт миокарда, миозиты, травмы).
Электронейромиография (ЭНМГ)
Для точной диагностики БАС необходимо проведение как игольчатой электромиографии, с использованием тонких игл, с помощью которых врач оценивает состояние электрической активности поврежденных и неповрежденных мышц, так и стимуляционной электронейромиграфии для исключения нарушения проведения по чувствительным волокнам, которые при БАС практически всегда остаются нетронутыми.
Транскраниальная магнитная стимуляция (ТМС)
Это новый метод, который может быть проведен одновременно с ЭНМГ. Он разработан для оценки состояния двигательных нейронов головного мозга. Результаты ТМС могут помочь в постановке диагноза.
Магнитно-резонансная томография (МРТ)
МРT способна выявить повреждения в головном и спинном мозге, вызванные инсультом, болезнью Альцгеймера, болезнью Паркинсона, рассеянным склерозом, опухолями и травмами спинного и головного мозга. В отдельных случаях при проведении МРТ выявляются признаки, характерные только для БДН/БАС – это свечение пирамидных путей, которые поражаются при этом заболевании.
Другие методы
Для верификации диагноза также необходимо проведение люмбальной пункции, оценки функции внешнего дыхания, проведение кардиореспираторного мониторинга. Данные исследования позволяют выявлять изменения, которые на ранних этапах не беспокоят пациента, однако вносят губительный вклад в прогрессирование заболевания.
Всегда следует помнить, что БАС – это болезнь исключения, и окончательный диагноз ставится только после полноценного качественного обследования пациента под наблюдением опытных неврологов, имеющих большой опыт работы с такими пациентами.
Цели терапии
В настоящее время не существует эффективных методов лечения данного заболевания. Поэтому терапия направлена на то, чтобы:
-
замедлить прогрессирование болезни и продлить период заболевания, при котором больной не нуждается в постоянном постороннем уходе;
-
уменьшить выраженность отдельных симптомов болезни и поддерживать стабильный уровень качества жизни.

Лечение
На сегодняшний день к неизлечимым заболеваниям относят и болезнь двигательного нейрона. Случаи выздоровления не были зафиксированы. Все средства современной медицины направлены на замедление развития дегенеративных процессов и улучшение состояния больного.
Единственным препаратом в лечении БДН, эффективность которого была доказана двумя независимыми клиническими исследованиями, является Рилузол (Рилутек). Его действие направлено на снижение уровня глутамата, который освобождается во время передачи нервных импульсов. Также положительное действие имеют препараты, связывающие свободные радикалы, и антиоксиданты.
Есть новые разработки в лечении болезни двигательного нейрона. Многие исследования доказывают иммунологическую природу развития патологии, однако, достоверно эффективные лекарства еще находятся на стадии усовершенствования.
К симптоматической терапии относят средства для улучшения состояния пациента. Применяют миорелаксанты для устранения болезненных спазмов мышц. Обязательно проводят поддержку дыхания с помощью неинвазивной вентиляции легких при помощи специальной маски.
При затруднениях при глотании кормление выполняется через зонд. Для этого составляется специальный рацион, удовлетворяющий все потребности организма в питательных веществах.
Прогноз заболевания неблагоприятный. В редких случаях больные живут до 10 лет. Обычно продолжительность жизни составляет 2-4 года после постановки диагноза.
В Юсуповской больнице предоставляют качественную паллиативную помощь больным с болезнью двигательного нерва. В отделении неврологии работает профессиональный медицинский персонал, который владеет спецификой оказания помощи подобным больным. В Юсуповской больницы созданы все условия для комфортного пребывания пациента с предоставлением ему всех необходимых медицинских услуг.
Общие представления
Потеря способности контролировать движения произвольной мускулатуры обычно описывается больными как «мышечная слабость» или как затруднения, которые можно интерпретировать как «потерю проворства». Диагностический подход к проблеме начинается с определения того, какая часть нервной системы поражена. Важно выяснить, возникает ли слабость от заболевания верхних мотонейронов (мотонейронов в коре мозга и их аксонов, опускающихся сквозь подкорковое белое вещество, внутреннюю капсулу, ствол мозга и далее по спинному мозгу) или же от заболевания «двигательной единицы» (нижних мотонейронов в вентральных рогах спинного мозга и их аксонов в спинномозговых корешках и периферических нервах, нервномышечной передачи и скелетной мускулатуры). [Каждая двигательная нервная клетка с помощью древовидных ветвлений концевых частей ее волокон контактирует со многими мышечными волокнами: вместе они образуют «двигательную единицу». — Прим. перев.
Общая характеристика: дисфункция верхних (центральных) мотонейронов — повышенный мышечный тонус (спастичность), повышенные глубокие сухожильные рефлексы, положительный симптом Бабинского.
Дисфункция нижних (периферических) мотонейронов: сниженный мышечный тонус, сниженные рефлексы, мышечная атрофия.
В табл. 12-1 представлены симптомы мышечной слабости и другие проявления, возникающие при поражении различных участков нервной системы. В табл.12-2 — наиболее частые причины мышечной слабости в связи с первичной локализацией поражения.
Оценка
Из анамнеза обращают внимание на скорость развития мышечной слабости, наличие нарушений чувствительности и других неврологических симптомов, информацию о применении лекарств, предрасполагающие факторы и семейный анамнез. При физикальном обследовании следует определить локализацию поражения в соответствии с критериями, изложенными ранее, и в табл. 12-1.
При поражении головного или спинного мозга для распознавания причин заболевания важны такие исследования, как КТ, МРТ, миелография, особенно при структурных и демиелинизирующих процессах. Люмбальная пункция диагностически значима при демиелинизирующих и инфекционных процессах; исследование сыворотки крови и мочи — при расстройствах питания и интоксикациях; биопсия пораженной ткани — для дифференциальной диагностики между опухолевым и инфекционным процессом.
Таблица 12-1 Клинические различия мышечной слабости при поражении различных отделов нервной системы
| Локализация поражения | Клинические признаки мышечной слабости | Сопутствующие симптомы |
| ВЕРХНИЙ МОТОНЕЙРОН (центральный паралич) | ||
| Кора головного мозга | Гемипарез (лицо и рука преимущественно, или нога преимущественно) | Односторонняя потеря чувствительности, судороги, гомонимная гемианопсия или квадрантная гемианопсия, афазия, апраксия |
| Внутренняя капсула | Гемипарез (лицо, рука, нога могут быть затронуты в равной степени) | Одностороннее снижение чувствительности, гомонимная гемианопсия или квадрантная гемианопсия |
| Ствол головного мозга | Гемипарез (рука и нога; лицо может быть совсем не вовлечено) | Головокружение, тошнота и рвота, атаксия и дизартрия, нарушение движений глазных яблок, нарушение черепной иннервации, нарушение сознания, синдром Горнера |
| Спииной мозг (поражение обеих половин) | Тетрапарез, если страдают верхние и средние шейные отделы; нижний парапарез, если страдают нижние шейные и грудные отделы | Уровень чувствительных расстройств; дисфункция прямой кишки и мочевого пузыря |
| Спинной мозг (поражение одной половины) | Гемипарез ниже уровня поражения (синдром Броун-Секара) | Потеря болевой чувствительности на противоположной стороне, тактильной и мышечно-суставной на стороне поражения ниже его уровня |
| «ДВИГАТЕЛЬНАЯ ЕДИНИЦА» (периферический паралич) | ||
| Стволовые и спинальные мотонейроны | Слабость мышц, иннервируемых мотонейронами двигательного ядра черепного нерва или переднего рога спинального сегмента | Фасцикуляции и фибрилляции страдающих мышц и их атрофия; нарушения чувствительности в соответствующих дерматомах |
Таблица 12-1 Клинические различия мышечной слабости при поражении различных отделов нервной системы
(продолжение)
| Локализация поражения | Клинические признаки мышечной слабости | Сопутствующие симптомы |
| Корешок спинного мозга | Слабость мышц, иннервируемых корешком черепного нерва или спинного мозга | Потеря чувствительности и корешковая боль в соответствующем дерматоме |
| Периферический нерв Полиневропатия | Периферическая мышечная слабость больше в стопах, чем в кистях; характерна симметрия | Периферические нарушения чувствительности, обычно в стопах в большей степени, чем в кистях |
| Мононевропатия | Мышечная слабость соответственно зоне иннервации отдельного нерва | Потеря чувствительности и боли в зоне иннервации отдельного нерва |
| Нервно-мышечное соединение | Патологическая мышечная утомляемость, часто в сочетании с нарушением зрения в виде диплопии и птоза | Нарушений чувствительности нет; рефлексы в норме |
| Мышцы | Слабость (чаще) проксимальных мышц | Нет нарушений чувствительности, при выраженном поражении — снижение рефлексов; может быть боль в мышцах при пальпации |
Разобраться в поражении «двигательной единицы» помогают электромиогра-фия и исследование скорости проведения импульса по нервным волокнам. В результате удается дифференцировать поражения различных компонентов «двигательной единицы». МРТ или миелография важны для оценки структурных причин заболеваний нервных корешков: исследование мочи и сыворотки крови для исключения системных поражений могут быть дополнены анализом спинномозговой жидкости; КК сыворотки — индикатор поражения мышечной ткани; биопсия нервной ткани используется редко, но биопсия мышечной ткани диагностически значима для многих мышечных заболеваний.
Таблица 12-2 Распространенные причины мышечной слабости
ВЕРХНИЕ МОТОНЕЙРОНЫ (ЦЕНТРАЛЬНОЕ ПОРАЖЕНИЕ)
Кора: ишемия, кровоизлияние, повреждение коры в результате интрацеребрального патологического процесса (первичный или метастатический рак, абсцесс); повреждение коры экстрацеребральным патологическим процессом (субдуральная гематома); дегенерация (боковой амиотрофический склероз).
Подкорковое белое вещество или внутренняя капсула: ишемия, кровоизлияние, повреждение ткани интрацеребральным патологическим процессом (первичный или метастатический рак, абсцесс); патоиммунный процесс (рассеянный склероз); инфекция (прогрессирующая многоочаговая лейкоэнцефалопатия).
Ствол головного мозга: ишемия, патоиммунный процесс (рассеянный склероз). Спинной мозг: компрессия (шейный спондилёз, метастазы рака, эпидуральный абсцесс); патоиммунный процесс (рассеянный склероз, поперечный миелит); инфекция (миелопатия при СПИДе, миелопатия, обусловленная HTLV-1, tabes dorsalis); дефицит питания (подострая комбинированная дегенерация).
«ДВИГАТЕЛЬНАЯ ЕДИНИЦА» (ПЕРИФЕРИЧЕСКОЕ ПОРАЖЕНИЕ)
Мотонейроны спинного мозга: дегенерация (боковой амиотрофический склероз);
инфекция (полиомиелит).
Спинномозговой корешок: компрессия (дегенеративное заболевание диска); патоиммунный процесс (синдром Гийена — Барре), инфекция (полирадикулопатия, ассоциированная со СПИДом, болезнь Лайма)
Периферический нерв: метаболическое поражение (сахарный диабет, уремия, порфи-рия); токсическое поражение (этанол, тяжелые металлы, обилие лекарств, дифтерия); дефицит питания (дефицит цианокобаламина); патоиммунный процесс (паранеопласти-ческий, парапротеинемия); инфекция (ассоциированная со СПИДом полиневропатия, множественная мононевропатия); компрессия нерва.
Нервно-мышечная передача: патоиммунный процесс (myasthenia gravis); токсическое воздействие (ботулизм, аминогликозиды).
Мышцы: воспаление (полимиозит, включая генерализованный миозит); дегенерация (мышечная дистрофия); токсическое поражение (глюкокортикоиды, этанол, азатиоприн); инфекция (трихинеллез); обменные поражения (гипотиреоз, периодический паралич); врожденные болезни (болезнь центрального стержня).
Двигательные расстройства
Двигательные расстройства часто делят на акинетические ригидные формы, при которых отмечаются мышечная скованность и замедленность движений, и гиперкинетические формы, когда выражены непроизвольные движения. В любом случае мышечная сила, как правило, сохраняется.
Большинство двигательных расстройств возникает в результате нарушения функциональной активности медиаторов в базальных ганглиях, патогенез может 6ып> различным. Наиболее частые причины: дегенеративные заболевания (врожденные или идиопатические), возможно, спровоцированные приемом лекарств, несостоятельность систем органов, инфекции ЦНС или ишемия базальных ганглиев. Ниже представлен краткий обзор клинических аспектов основных категорий двигательных расстройств.
Брадикинезия
Неспособность больного начинать движение или легко и быстро выполнять обычные произвольные движения. Отмечаются замедленность движений, уменьшение количества автоматических движений, таких как размахивание руками при ходьбе и моргание. Это обычно указывает на болезнь Паркинсона.
Тремор
Ритмичные колебания части тела относительно фиксированной точки, обычно наблюдается тремор дистальных отделов конечностей и, реже, головы, языка или нижней челюсти. Тремор можно подразделить на виды в зависимости от локализации и амплитуды движений. Чаще всего встречается крупноразмашистый тремор в покое — 4-5 мышечных сокращений в 1 с, что является признаком болезни Паркинсона; выраженный постуральный (тонический) тремор с периодичностью 8-10 мышечных сокращений в 1 с может быть гипертрофированным вариантом физиологического тремора или признаком эссенциального наследственного тремора, характерного для нескольких членов одной семьи. Для лечения последнего применяют пропранолол (анаприлин) или примидон (гексамидин).
Астериксис
Быстрые аритмичные движения, прерывающие фоновые произвольные сокращения мускулатуры, обычно это быстрые сгибания и разгибания кистей вытянутых вперед рук. Этот «печеночный хлопок» (при печеночной недостаточности) может наблюдаться также при лекарственной энцефалопатии, при несостоятельности некоторых систем органов или инфекции ЦНС. Лечение должно быть направлено на заболевание, лежащее в основе тремора.
Миоклонус
Кратковременные аритмичные мышечные сокращения или подергивания. Как и астериксис, обычно является признаком рассеянной энцефалопатии, иногда отмечается после кратковременной остановки сердца, когда распространенная гипоксия мозга вызывает многоочаговый миоклонус. Эффективно лечение следующими препаратами: клоназепам, вальпроат, баклофен.
Дистония
Непроизвольная длительная поза или малоизменяемые фиксированные патологические позы. Они часто нелепы, вычурны, с насильственным сгибанием или разгибанием в отдельных суставах. Дистонии носят генерализованный или очаговый характер (спастическая кривошея, блефароспазм). Симптоматическое лечение проводят высокими дозами антихолинергических препаратов, бензодиазепинов, бакло-фена и антиконвульсантов. Локальные инъекции ботулотоксина эффективны при некоторых очаговых дистониях.
Хореоатетоз
Комбинация хореи (быстрые, порывистые движения) и атетоза (медленные судорожные движения). Эти два типа патологических движений сосуществуют, хотя один из компонентов может быть выражен в большей степени. Хореические симптомы преобладают при непроизвольных движениях ревматической (болезнь Сиден-гама) хореи и болезни Гентингтона. Атетоз доминирует в картине некоторых форм церебрального паралича. Длительный прием нейролептиков может вести к медленной дискинезии, в симптоматике которой хореоатетоз охватывает мускулатуру щек, языка и нижней челюсти. В лечении применяют бензодиазепины, резерпин, низкие дозы нейролептиков, хотя результаты часто разочаровывают.
Тики
Стереотипные, бессмысленные движения, как моргание, чихание или покашливание. Синдром Жилля де ля Туретта встречается редко, протекает тяжело. Его клиническая картина включает моторные тики (конвульсии лица, шеи, плечей), головные тики (хрюканье, произнесение слов), «поведенческие тики» (копролалия, эхолалия). Причины синдрома неизвестны. Лечение галоперидолом обычно снижает частоту и выраженность проявлений синдрома.
Боковой (латеральный) амиотрофический склероз (БАС) (также известен как болезнь моторных нейронов, Мотонейронная болезнь, болезнь Шарко, в англоязычных странах — болезнь Лу Герига) — прогрессирующее, неизлечимое дегенеративное заболевание центральной нервной системы, при котором происходит поражение как верхних (моторная кора головного мозга), так и нижних (передние рога спинного мозга и ядра черепно-мозговых нервов) двигательных нейронов, что приводит к параличам и последующей атрофии мышц.
Общие сведения.
Болезнь известна не так давно. Впервые описана Жан-Мартеном Шарко в 1869г. По статистике выявляется у 2-5 человек на 100 000 населения в год, что говорит о том, что данная патология относительно редко встречается. Всего в мире насчитывается около 70 тысяч больных боковым амиотрофическим склерозом. Обычно заболевание заявляет о себе у людей старше 50 лет.
Совсем недавно было высказано мнение, что случаи бокового амиотрофического склероза чаще регистрируются у высокоинтеллектуальных людей, профессионалов в своем деле, а также у спортсменов-атлетов, которые на протяжении всей жизни отличались крепким здоровьем.
В 90% случаев БАС носит спорадический, а в 10% — семейный или наследственный характер как с аутосомно-доминантным (преимущественно), так и с аутосомно-рецессивным типами наследования. Клинические и патоморфологические характеристики семейного и спорадического БАС практически идентичны.
Точная этиология БАС неизвестна.
Патогенез.
Сущность болезни заключается в дегенерации двигательных нейронов, т.е. под воздействием ряда причин запускается процесс разрушения нервных клеток, ответственных за сокращения мышц. Этот процесс затрагивает нейроны коры больших полушарий, ядер головного мозга и нейроны передних рогов спинного мозга. Двигательные нейроны погибают, а их функции никто больше не выполняет. Нервные импульсы к мышечным клеткам больше не поступают. И мышцы слабеют, развиваются парезы и параличи, атрофия мышечной ткани.
Если в основе бокового амиотрофического склероза лежит мутация в гене супероксиддисмутазы-1, то процесс выглядит примерно следующим образом. Мутантная супероксиддисмутаза-1 накапливается в митохондриях двигательных нейронов (в энергетических станциях клетки). Это «мешает» нормальному внутриклеточному транспорту белковых образований. Белки соединятся друг с другом, как бы слипаются, и это запускает процесс дегенерации клетки.
Если причиной становится избыток глутамата, то механизм запуска разрушения двигательных нейронов выглядит так: глутамат открывает каналы в мембране нейронов для кальция. Кальций устремляется внутрь клеток. Избыток кальция, в свою очередь, активирует внутриклеточные ферменты. Ферменты как бы «переваривают» структуры нервных клеток, при этом образуется большое количество свободных радикалов. И эти свободные радикалы повреждают нейроны, постепенно приводя к их полному разрушению.
Предполагается, что роль других факторов в развитии БАС также заключается в запуске свободнорадикального окисления.
Классификация БАС, формы:
- пояснично-крестцовая;
- шейно-грудная;
- бульбарная: при поражении периферического мотонейрона в стволе головного мозга;
- высокая: при поражении центрального мотонейрона.
Общими симптомами, характерными для любой из форм бокового амиотрофического склероза, являются:
- сугубо двигательные нарушения;
- отсутствие чувствительных расстройств;
- отсутствие расстройств со стороны органов мочеиспускания и дефекации;
- неуклонное прогрессирование болезни с захватом новых мышечных массивов вплоть до полной обездвиженности;
- наличие периодических болезненных судорог в пораженных частях тела, их называют крампи.
Клиника
Начальные проявления заболевания:
•слабость в дистальных отделах рук, неловкость при выполнении тонких движений пальцами, похудание в кистях и фасцикуляции (мышечные подергивания)
•реже заболевание дебютирует слабостью в проксимальных отделах рук и плечевом поясе, атрофиями в мышцах ног в сочетании с нижним спастическим парапарезом
•возможно также начало заболевания с бульбарных расстройств – дизартрии и дисфагии (25% случаев)
•крампи (болезненные сокращения, спазмы мышц), нередко генерализованные, встречаются практически у всех больных БАС, и нередко являются первым признаком заболевания
Для БАС в большинстве случаев характерна асимметричность симптоматики.
Пояснично-крестцовая форма:
При этой форме заболевания возможно два варианта:
- болезнь начинается только с поражения периферического мотонейрона,расположенного в передних рогах пояснично-крестцового отдела спинного мозга. В этом случае у больного развивается мышечная слабость в одной ноге, затем она появляется и в другой, снижаются сухожильные рефлексы (коленный, ахиллов), снижается тонус мышц в ногах, постепенно формируются атрофии (это выглядит как похудение ног, как бы «усыхание»). Одновременно с этим в ногах наблюдаются фасцикуляции – непроизвольные мышечные подергивания с небольшой амплитудой («волны» мышц, мышцы «шевелятся»). Затем в процесс вовлекаются мышцы рук, в них также снижаются рефлексы, образуются атрофии. Процесс идет выше – вовлекается бульбарная группа мотонейронов. Это приводит к появлению таких симптомов, как нарушение глотания, смазанность и нечеткость речи, гнусавый оттенок голоса, истончение языка. Возникают поперхивания при приеме пищи, начинает отвисать нижняя челюсть, появляются проблемы с жеванием. На языке также бывают фасцикуляции;
- в начале болезни выявляются признаки одновременного поражения центрального и периферического мотонейронов, обеспечивающих движения в ногах. При этом слабость в ногах сочетается с повышением рефлексов, повышением мышечного тонуса, атрофиями мышц. Появляются патологические стопные симптомы Бабинского, Гордона, Шеффера, Жуковского и др. Затем аналогичные изменения возникают в руках. Потом вовлекаются и мотонейроны головного мозга. Появляются нарушения речи, глотания, жевания, подергивания в языке. Присоединяются насильственный смех и плач.
Шейно-грудная форма:
Также может дебютировать двумя способами:
- поражение только периферического мотонейрона – появляются парезы, атрофии и фасцикуляции, снижение тонуса в одной кисти. Через пару месяцев те же симптомы возникают и в другой кисти. Кисти рук приобретают вид «обезьяньей лапы». Одновременно в нижних конечностях выявляют повышение рефлексов, патологические стопные знаки без атрофий. Постепенно мышечная сила снижается и в ногах, в процесс вовлекается бульбарный отдел головного мозга. И тогда присоединяются нечеткость речи, проблемы с глотанием, парезы и фасцикуляции языка. Слабость в мышцах шеи проявляется свисанием головы;
- одновременное поражение центрального и периферического мотонейронов. В руках одновременно присутствуют атрофии и повышенные рефлексы с патологическими кистевыми признаками, в ногах – повышение рефлексов, снижение силы, патологически стопные симптомы при отсутствии атрофий. Позже поражается бульбарный отдел.
Бульбарная форма:
- При этой форме заболевания первыми симптомами при поражении периферического мотонейрона в стволе мозга становятся расстройства артикуляции, поперхивание при приеме пищи, гнусавость голоса, атрофия и фасцикуляции языка. Движения языка затруднены. Если поражен и центральный мотонейрон, то к этим симптомам присоединяются и повышение глоточного и нижнечелюстного рефлексов, насильственный смех и плач. Повышается рвотный рефлекс.
В руках по мере прогрессирования болезни формируется парез с атрофическими изменениями, повышением рефлексов, повышением тонуса и патологическими стопными признаками. Аналогичные изменения возникают и в ногах, но несколько позже.
Высокая форма:
Это разновидность бокового амиотрофического склероза, когда заболевание протекает с преимущественным поражением центрального мотонейрона. При этом во всех мышцах туловища и конечностей формируются парезы с повышением мышечного тонуса, патологическими симптомами.
Бульбарная и высокая формы БАС являются прогностически неблагоприятными. Больные с таким началом заболевания имеют меньшую продолжительность жизни по сравнению с шейно-грудной и пояснично-крестцовой формами. Какими бы ни были первые проявления заболевания, оно неуклонно прогрессируют.
Парезы в различных конечностях приводят к нарушению способности самостоятельно передвигаться, обслуживать себя. Вовлечение в процесс дыхательной мускулатуры приводит вначале к появлению одышки при физической нагрузке, затем одышка беспокоит уже в покое, появляются эпизоды острой нехватки воздуха. В терминальных стадиях самостоятельное дыхание просто невозможно, больным требуется постоянная искусственная вентиляция легких.
Продолжительность жизни больного БАС составляет по разным данным от 2 до 12 лет, однако более 90% больных умирают в течение 5 лет от момента постановки диагноза. В терминальную стадию болезни больные полностью прикованы к постели, дыхание поддерживается с помощью аппарата искусственной вентиляции легких. Причиной гибели таких больных может стать остановка дыхания, присоединение осложнений в виде пневмонии, тромбоэмболии, инфицирования пролежней с генерализацией инфекции.
Диагностика:
Среди параклинических исследований наиболее существенное диагностическое значение имеет электромиография. Выявляется распространенное поражение клеток передних рогов (даже в клинически сохранных мышцах) с фибрилляциями, фасцикуляциями, позитивными волнами, изменениями потенциалов двигательных единиц (увеличивается их амплитуда и длительность) при нормальной скорости проведения возбуждения по волокнам чувствительных нервов. Содержание КФК в плазме может быть незначительно повышено
Боковой амиотрофический склероз нужно заподозрить:
•при развитии слабости и атрофий, а возможно и фасцикуляций (мышечных подергиваний) в мышцах кисти
•при похудания мышц тенара одной из кистей с развитием слабости аддукции (приведения) и оппозиции большого пальца (обычно асимметрично)
•при этом наблюдается затруднение при схватывании большим и указательным пальцами, затруднения при подбирании мелких предметов, при застегивании пуговиц, при письме
•при развитии слабости в проксимальных отделах рук и плечевом поясе, атрофий в мышцах ног в сочетании с нижним спастическим парапарезом
•при развитии у пациента дизартрии (нарушений речи) и дисфагии (нарушений глотания)
•при появлении у пациента крампи (болезненных мышечных сокращений)
Диагностические критерии БАС:
- Симптомы поражения нижнего моторного нейрона (включая ЭМГ-подтверждение в клинически сохранных мышцах).
- Симптомы поражения верхнего моторного нейрона
- Прогрессирующее течение
Критерии исключения БАС
Для диагностики бокового амиотрофического склероза необходимо отсутствие:
•сенсорных расстройств, в первую очередь выпадений чувствительности (возможны парестезии и боли)
•тазовых расстройств — нарушений мочеиспускания и дефекации (их присоединение возможно на конечных стадиях заболевания)
•зрительных нарушений
•вегетативных нарушений
•болезни Паркинсона
•деменции альцгеймеровского типа
•синдромов, похожих на БАС
Критерии подтверждения БАС:
Диагноз БАС подьверждается:
- Фасцикуляциями в одной и более областях
- ЭМГ-признаки нейронопатии
- Нормальной скоростью проведения возбуждения по моторным и сенсорным волокнам (дитсальные моторные латенции могут быть увеличенными)
- Отсутствием блока проведения
Дифференциальный диагноз БАС (синдромы похожие на БАС):
•Спондилогенная шейная миелопатия.
•Опухоли краниовертебральной области и спинного мозга.
•Краниовертебральные аномалии.
•Сирингомиелия.
•Подострая комбинированная дегенерация спинного мозга при недостаточности витамина В12.
•Семейный спастический парапарез Штрюмпеля.
•Прогрессирующие спинальные амиотрофии.
•Постполиомиелитический синдром.
•Интоксикации свинцом, ртутью, марганцем.
•Недостаточность гексозаминидазы типа А у взрослых при ганглиозидозе GM2.
•Диабетическая амиотрофия.
•Мультифокальная моторная невропатия с блоками проведения.
•Болезнь Крейцтфельдта-Якоба.
•Паранеопластический синдром, в частности при лимфогранулематозе и злокачественной лимфоме.
•Синдром БАС при парапротеинемии.
•Аксональная нейропатия при болезни Лайма (Лайм-боррелиозе).
•Синдром Гийена-Барре.
•Миастения.
•Рассеянный склероз
•Эндокринопатии (тиреотоксикоз, гиперпаратиреоз, диабетическая амиотрофия).
•Доброкачественные фасцикуляции, т.е. фасцикуляции, продолжающиеся годами без признаков поражения двигательной системы.
•Нейроинфекции (полиомиелит, бруцеллез, эпидемический энцефалит, клещевой энцефалит, нейросифилис, болезнь Лайма).
•Первичный боковой склероз.
Диагностические исследования при синдроме БАС.
Для уточнения диагноза и проведения дифференциального диагноза при синдроме БАС рекомендутся следующее обследование больного:
Анализ крови (СОЭ, гематологические и биохимические исследования)
Рентгенография органов грудной клетки
ЭКГ
Исследование функций щитовидной железы
Определение содержания витамина В12 и фолиевой кислоты в крови
Креатинкиназа в сыворотке
ЭМГ
МРТ головного мозга и при необходимости, спинного мозга
Люмбальная пункция.
Лечение
Эффективного лечения заболевания не существует. Единственный препарат, ингибитор высвобождения глутамата рилузол (Рилутек), отодвигает летальный исход на 2 – 4 месяца. Его назначают по 50 мг два раза в день.
Основу лечения составляет симптоматическая терапия:
•Лечебная гимнастика.
•Физическая активность. Пациент должен по мере своих возможностей поддерживать физическую активность По мере прогрессирования заболевания возникает необходимость в кресле-каталке и других специальных приспособлениях.
•Диета. Дисфагия создаёт опасность попадания пищи в дыхательные пути • Иногда возникает необходимость в питании через зонд или в гастростомии.
•Применение ортопедических приспособлений: шейного воротника, различных шин, устройств для захвата предметов.
•При крампи (болезненным мышечных спазмах): карбамазепин (Финлепсин, Тегретол) и/или витамин Е, а также препараты магния, верапамил (Изоптин).
•При спастичности: баклофен (Баклосан), Сирдалуд, а также клоназепам.
•При слюнотечении атропин, или гиосцин (Бускопан).
•При невозможности приема пищи вследствие нарушения глотания накладывают гастростому или вводят назогастральный зонд. Раннее проведение чрезкожной эндоскопической гастростомии продлевает жизнь пациентов в среднем на 6 месяцев.
•При болевых синдромах используют весь арсенал аналгетиков. В том числе на конечных стадиях наркотические аналгетики.
•Иногда некоторое временное улучшение приносят антихолинэстеразные препараты (неостигмина метилсульфат — прозерин).
•Церебролизин в высоких дозах (10-30 мл в/в капельно 10 дней повторными курсами). Существует ряд небольших исследований, показывающих нейропротективную эффективность церебролизина при БАС.
•Антидепрессанты: Серталин или Паксил или Амитриптилин (часть больных БАС предпочитает именно его как раз из-за побочных действий – он вызывает сухость во рту, соответственно уменьшает гиперсаливацию (слюнотечение), часто мучающую больных БАС).
•При появлении дыхательных нарушений: искусственная вентиляция легких в условиях стационаров, как правило, не проводится, но некоторые больные приобретают портативные приборы ИВЛ и проводят ИВЛ в домашних условиях.
•Ведутся разработки к применению гормона роста, нейротрофических факторов при БАС.
•Последнее время активно ведутся разработки лечения стволовыми клетками. Этот метод обещает быть перспективным, но все же пока находится на стадии научных экспериментов.
Прогноз
•Боковой амиотрофический склероз является фатальным заболеванием. Средняя продолжительность жизни больных БАС 3 – 5 лет, тем не менее, 30% больных живут 5 лет, а около 10 – 20% живут более 10 лет от начала заболевания.
•Неблагоприятные прогностические признаки – пожилой возраст и бульбарные нарушения (после появления последних больные живут не более 1 – 3 лет).
Специфическая профилактика отсутствует.
Дата публикации 28 мая 2020Обновлено 3 ноября 2022
Определение болезни. Причины заболевания
Боковой амиотрофический склероз (БАС, англ. ALS) — это неуклонно прогрессирующее заболевание, характеризующееся гибелью (дегенерацией) моторных нейронов, что приводит к нарушению движения мышц, в том числе дыхательных, вплоть до параличей и аторфии мышц.

Чтобы понять суть заболевания, необходимо коснуться строения и функций головного и спинного мозга. В структуре спинного мозга на всём его протяжении и частично в стволе головного мозга существуют клетки, посылающие нервный импульс прямо к мышечным волокнам. Они называются нижние мотонейроны, так как своими импульсами приводят мышцы в движение. Группируясь в передней части поперечного среза спинного мозга, нижние мотонейроны образуют так называемый «передний рог».

Также спинной мозг выполняет функцию соединяющего нервного «кабеля» между головным мозгом и частями тела. В норме спинной мозг подчиняется головному мозгу. Это означает, что, если импульс от головного мозга укажет мышце поднять руку, а импульс спинного мозга укажет опустить, то рука поднимется.
Головой мозг имеет сложное строение, но в данной статье нужно уделить внимание верхним мотонейронам. Они группируются в области, которая отвечает на исходную генерацию двигательных импульсов в организме — коре больших полушарий и стволе мозга. Импульсы, в свою очередь, идут по спинному мозгу вниз к вышеупомянутым нижним мотонейронам, а оттуда к мышцам, приводя их в движение. Именно этот путь поражается при боковом амиотрофическом склерозе.

БАС был впервые описан в 1869 году. В литературе можно встретить следующие синонимы БАС: болезнь моторных нейронов, мотонейронная болезнь, болезнь Шарко (по фамилии врача-первооткрывателя, хотя открыл это заболевание Чарльз Белл [1]), болезнь Лу Герига (по имени знаменитого бейсболиста, умершего от БАС в 1938 году). Сейчас многие называют ее болезнью Стивена Хокинга, потому что он прожил с БАС более 50 лет.
")
Боковой амиотрифический склероз — представитель группы болезней двигательного нейрона, т. е. болезней мотонейронов. Кроме БАС, в эту группу включены: первичный латеральный склероз, прогрессирующая мышечная атрофия и прогрессирующий бульбарный паралич.
Учёные выявили две причины БАС: 90-95 % — спорадическая причина (проще говоря случайность) и 5-10 % — генетически наследуемая мутация [2].
В ходе многолетних исследований были найдены негенетические модифицируемые факторы образа жизни, предрасполагающие к БАС:
- Курение сигарет является наиболее постоянным негенетическим фактором риска для БАС [8].
- Контакт с сельскохозяйственными удобрениями: пестицидами, гербицидами, инсектицидами и формальдегидом [8]. Стоит упомянуть, что формальдегид в повседневной жизни встречается в табачном дыме, поэтому пассивного курения тоже стоит избегать.
- Свинец, выделяемый в окружающую среду в профессиональной деятельности, например, при сварке металлов [10].
- Микротравмы головы в профессиональном спорте [9]. Это значит, что Лу Геринг, будучи бейсболистом, вероятно спровоцировал своей профессией БАС и раннюю смерть через 2 года.
- Употребление грибов или растений в пищу. Известны два случая «вспышки» БАС в определённых местностях. Например, в тихоокеанском городе Гуам население традиционно употребляло в пищу и лечилось саго — крахмалом саговой пальмы. Содержащийся в нём токсин привёл к тому, что БАС стал встречаться в 50 раз чаще [17]. Такую форму БАС назвали болезнью Гуам, но она встречается и в других странах [18]. Ещё один случай внезапной «вспышки» БАС произошёл в Альпах: местные жители стали есть ложные сморчки и токсины этих грибов привели к генетическим мутациям [19].
Обобщая вышесказанное, можно сказать, что БАС развивается в результате комбинированного воздействия генов, факторов окружающей среды и образа жизни. Эта модель (ген-время-среда) предполагает, что развитие БАС является многоэтапным процессом, в котором генетические дефекты являются лишь одним из нескольких этапов, в конечном итоге приводящих к БАС.
В нашей стране проживает примерно 10-12 тысяч пациентов с БАС. Заболевание чаще встречается у мужчин, чем у женщин (в 1,5 раза), а возрастной диапазон — от 20 до 80 лет. Скорость его прогрессирования прямо пропорциональна возрасту. Наличие генетической мутации (у 5-10 % заболевших) также увеличивает скорость развития тяжёлых симптомов [14].
![]()
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением — это опасно для вашего здоровья!
Симптомы бокового амиотрофического склероза
Начальные симптомы БАС варьируют у пациентов в зависимости от степени поражения верхних и нижних двигательных нейронов и от того, какие участки тела вовлечены. Поэтому особенность бокового амиотрофического склероза заключается в том, что врачи часто вынуждены наблюдать за развитием жалоб в течение нескольких месяцев, прежде чем поставить верный диагноз (например, определить тип заболевания двигательного нейрона или тип БАС)
В классическом варианте болезнь начинается с поражения одного из отделов центральной нервной системы: ствол головного мозга, шейный, грудной или поясничный отдел спинного мозга. В зависимости от уровня будут поражаться (слабеть, истончаться, непроизвольно подёргиваться) определённые группы мышц.

| Уровень поражения нижнего мотонейрона | Ствол головного мозга | Шейный отдел спинного мозга | Грудной отдел спинного мозга | Поясничный отдел спинного мозга |
|---|---|---|---|---|
| Группы мышц, которые поражаются | Мыщцы лица, мягкого нёба, язык, мышцы гортани и глотки |
Мышцы шеи, рук | Мышцы спины, живота, диафрагмы |
Мышцы спины, живота, ног |
Подёргивание мышц при БАС имитирует судороги мышц после сильной физической нагрузки. Стоит отметить, что от слабости мышц при БАС не удаётся избавиться с помощью физических упражнений. Это связано с необратимым прогрессированием болезни [12].
По мере развития заболевания будут наблюдаться признаки поражения верхнего мотонейрона, а также постепенное присоединение соседних отделов спинного мозга.
Признаки поражения верхнего мотонейрона:
- Усиление нижнечелюстного рефлекса.
- Эмоциональная нестабильность (приступы неконтролируемого смеха и/или плача).
- Непроизвольный спазм (тризм) жевательных мышц и невозможность открыть рот.
- Спазм мышц гортани.
- Повышение сухожильных рефлексов в руках и ногах (вызываются ударами молоточка по сухожилиям рук и ног на приёме у невролога).
- Тугоподвижность и спастичность мышц.
- Патологические рефлексы. Многие из этих рефлексов есть у новорождённых детей, но в течение следующего года по мере созревания коры полушарий они угасают.
- рефлексы орального автоматизма — патологические рефлексы на лице, например хоботковый (вытягивание губ вперёд при постукивании молоточком по верхней или нижней губе) или сосательный (в случае прикосновения к губам наблюдаются сосательные движения);
- патологические кистевые и стопные рефлексы, например симптом Бабинского (патологический подошвенный разгибательный рефлекс). Он заключается в разгибании большого пальца и разведении других пальцев ноги («знак веера») в ответ на штриховое раздражение наружного края подошвы. В норме наблюдается сгибание пальцев ног.

Также среди симптомов можно отметить мышечные подёргивания, болезненные спазмы в мышцах, слабость в тех или иных группах мышц, которые не проходят, а наоборот, нарастают после целенаправленных силовых занятий в тренажёрном зале. Характерно похудание и скованность тех или иных мышц, нарушения мелких движений в кистях или движений верхнего плечевого пояса, нарушения ходьбы, речи, глотания, слюноотделения (из-за нарушения глотательной функции слюна скапливается во рту). Иногда возникают эпизоды острой нехватки воздуха, в том числе во время приёма пищи (не путать с эпизодами панических атак), нарушения чиханья, откашливания, одышка при физической нагрузке или без неё, снижение массы тела, двоение в глазах при взгляде прямо или в ту или иную сторону, безболевые ожоги, чувство ползания мурашек по телу, нарушение мочеиспускания и/или стула [3].
Нужно отметить, что не все из перечисленных признаков могут наблюдаться у людей с БАС, так как клиническая картина заболевания всегда индивидуальна [12].
Патогенез бокового амиотрофического склероза
Патогенез заболевания чётко не определён. Примерная схема такова: ген в хромосоме (например ген супероксиддисмутазы-1, находящийся на 21-й хромосоме) «ломается» по неизвестным причинам. В результате синтезируются неправильные белки, которые мешают нормальной работе клеток нервной системы. Нервные клетки из-за этой поломки погибают, но процесс переработки мёртвых клеток (процесс аутофагии) также по какой-то причине не происходит. Из-за этого мёртвые клетки не распадутся на составляющие, а значит не создают «строительного материала» для новых клеток.
Параллельно на этом фоне в огромных количествах начинает синтезироваться глутамат (глутамат натрия — это почти основное «топливо» нервной системы, которое синтезируется самими нейронами, чтобы передавать информацию друг другу). Повышение возбудимости здоровых нейронов из-за высокой концентрации глутамата компенсирует «силы» погибших нейронов. Однако с ростом глутаминовой кислоты возбуждение нейронов продолжает возрастать, происходит отравление нервной ткани глутаматом и патологический круг замыкается — организм вредит сам себе [3].
Все это соотносится с клиникой. Повышенная активность здоровых нейронов вызывает подёргивание мышц. Вскоре из-за скопления мёртвых клеток и отравления глутаматом нейроны погибают, происходит денервация мышц (разобщение связей мышц с нервной системой), в результате чего те слабеют и истончаются.
Классификация и стадии развития бокового амиотрофического склероза
Единой классификации БАС не существует, поскольку не существует единства представлений о его патогенезе. Рассмотрим Североамериканскую классификацию, которая близка к отечественной. Согласно этой классификация, БАС делится на две большие группы в зависимости от причин: семейный и спорадический (случайный).
Спорадический БАС делится на группы относительно уровня поражения моторных нейронов двигательного пути в дебюте заболевания. БАС может впервые проявиться на уровне ствола мозга, шейного, грудного (в том числе в виде слабости диафрагмальных мышц), поясничного отдела спинного мозга или на всем протяжении пути единовременно.
Семейная группа делится на виды в зависимости от типа мутации конкретного гена супероксиддисмутазы-1 (СОД-1) или мутаций в других хромосомах (около 10 других известных генов).
Североамериканская классификация БАС
Спорадический (случайный) БАС
- Классический БАС. Дебюты:
- бульбарный;
- шейный;
- грудной;
- поясничный;
- диффузный;
- респираторный.
- Прогрессирующий бульбарный паралич.
- Прогрессирующая мышечная атрофия.
- Первичный латеральный (боковой) склероз.
Семейный БАС
- Аутосомно-доминантный:
- ассоциированный с мутациями СОД-1;
- без мутации СОД-1 (мутации других генов, генетический дефект не известен).
- Аутосомно-рецессивный:
- ассоциированный с мутациями СОД-1;
- мутации других генов.
- Западно-тихоокеанский комплекс БАС-паркинсонизм-деменция.
В первую группу кроме классического БАС входят три патологии: первичный латеральный склероз; прогрессирующая мышечная атрофия; прогрессирующий бульбарный паралич. В нашей стране их считают отдельными заболеваниями и объединяют в группу болезней двигательного нейрона. В США данные патологии рассматривают как варианты проявления БАС. Несмотря на разницу классификаций, проявления этих заболеваний будут одинаковыми в любой стране.
- Классическая форма БАС имеет признаки поражения и верхнего и нижнего мотонейрона: слабость, истощение, подёргивание, спастичность мышц и др. (см. раздел «Симптомы»).
- Прогрессирующий бульбарный паралич характеризуется поражением нижнего мотонейрона на уровне ствола мозга. Он манифестирует нарушением речи и глотания, возникают затруднения при жевании, разговоре, появляются гнусавость голоса, снижение рвотного рефлекса и слабость мимической мускулатуры, языка и мягкого нёба. В последующем присоединяются расстройства движения в конечностях, как при БАС.
- Первичный латеральный склероз проявляется признаками поражения исключительно верхнего мотонейрона в головном мозге: мышечная слабость, скованность (спастичность) мышц.
- Прогрессирующая мышечная атрофия, когда наблюдаются симптомы поражения только нижнего мотонейрона в спинном мозге (слабость, атрофия, подёргивание мышц, усталость, судороги, утрата рефлексов).
Осложнения бокового амиотрофического склероза
- Тугоподвижность, слабость конечностей, вплоть до полной парализации из-за постепенного отмирания верхнего мотонейрона.
- Деформация стоп, спастическая контрактура кисти, плечелопаточный периартроз из-за истончения, слабости мышц и невозможности поддерживать форму суставов.

- Трудность удерживания шеи в вертикальном положении из-за слабости мышц.
- Нарушения ходьбы, вплоть до полной обездвиженности в процессе развития заболевания из-за слабости мышц ног.
- Тромбозы глубоких вен нижних конечностей из-за длительного постельного режима по мере прогрессирования заболевания.

- Слюнотечение из-за нарушения глотательной функции.
- Нарушение чёткости произношения слов, а затем отсутствие речи из-за снижения силы в мышцах лица.
- Трудность проглатывания пищи, ощущение комка в горле, поперхивания, вплоть до невозможности проглотить жидкую и твёрдую пищу.
- Синдром обструктивного апноэ во время сна (непроизвольная задержка дыхания во сне).
- Дыхательные нарушения, начиная с одышки и заканчивая смертельной остановкой дыхания.
Диагностика бокового амиотрофического склероза
При подозрении на болезнь двигательного нейрона пациент сначала отвечает на стандартные вопросы невролога о жалобах, развитии болезни, сопутствующих заболеваниях (стоит обратить внимание на онкологию и инфекции, так как они могут стать причиной ложноположительного БАС-симптома, а не самой болезни). Врач также спрашивает о болезнях родственников, стране рождения, о мочеиспускании и дефекации.
Невролог должен осмотреть мышцы тела, оценить их силу, тонус, проверить рефлексы с помощью молоточка, оценить чувствительность кожи при помощи иголки и камертона. При подозрении на БАС мышцы непроизвольно сокращаются и истончаются. В них теряется сила, даже если пациент занимается физическими упражнениями для их укрепления. Также врач может задать вопросы, похожие на определение IQ. При снижении интеллектуальных способностей можно заподозрить некий патологический процесс в коре головного мозга, где находится верхний мотонейрон.
Однако первым критерием подтверждения БАС является наличие мутации генов (например, мутации в супероксиддисмутазе-1), выявленное при генетической экспертизе. Тогда ставится семейная форма БАС. Если гены не «поломаны», то ставится спорадическая (случайная) форма БАС, возникшая из-за факторов риска. При этом главным инструментом в диагностике любых формах БАС является электромиография (ЭМГ).
Прибор ЭМГ в прямом смысле бьёт пациента током, чтобы посчитать скорость проведения электрического импульса по нервам руки или ноги к нижнему мотонейрону в спинном мозге. В случае БАС велика вероятность повторных ЭМГ с целью фиксирования изменений возбудимости нейронов во времени. Если вспомнить патогенез, то в начале заболевания из-за глутамата натрия нервы перевозбудимы, а затем они все больше и больше отмирают, следовательно, возбуждение на ЭМГ будет уменьшаться. Вспомогательным критерием является прогрессирующее распространение симптомов в пределах одной или нескольких областей иннервации, что выявляют при наблюдении за больным в течение нескольких месяцев, иногда на протяжении года-двух [3].

Диагноз бокового амиотрофического склероза невозможно достоверно поставить с первого посещения невролога только после одного исследования ЭМГ. Иногда с целью уточнения заболевания или формы БАС врач дополнительно может назначить:
- клинический анализ крови (содержание гемоглобина, лейкоцитарная формула, СОЭ);
- биохимический анализ крови (общий белок, белковые фракции, мочевина, креатинин, билирубин, КФК);
- серологические анализы для исключения ложноположительного БАС-синдрома, связанного с инфекциями, например, могут назначить реакцию Вассермана для исключения сифилиса или анализ на антитела к ВИЧ, боррелиям и др.;
- иногда в условиях стационара неврологии могут сделать спинномозговую пункцию, чтобы исследовать ликвор на нейроинфекции;
- МРТ головного и спинного мозга.
Функциональную способность пациентов оценивают в баллах по специальной шкале. В соответствии с этой шкалой максимальный балл 48 соответствует полной функциональной способности больного, а минимальный балл 0 соответствует максимально выраженной инвалидизации пациента.
Скорость прогрессирования заболевания рассчитывают по формуле: в числителе — 48 минус баллы по функциональной шкале, в знаменателе — длительность заболевания в месяцах. Значение менее 0,45 условной единицы означает медленное прогрессирование болезни, значение от 0,45 до 0,54 — быстрое, от 0,55 и более — стремительное [16].
Как устанавливают диагноз
Для постановки диагноза «БАС» врачи используют критерии El Escorial. Они включают комбинацию поражений верхнего и нижнего мотонейрона на разных уровнях спинного мозга: шея, грудной и поясничный отдел. Их необходимо подтвердить клинически и с помощью ЭМГ. Также нужно убедиться, что состояние пациента ухудшается в течение последних 6 месяцев [20].
К критериям, исключающим БАС, относятся:
- нарушение поверхностной и глубокой чувствительности, т. е. не только кожи, но и связок, мышц, костей и др.;
- различные поражения вегетативной нервной системы, из-за которых нарушается работа внутренних органов и систем;
- тазовые нарушения, например проблемы с мочеиспусканием;
- зрительные нарушения, например выпадения полей зрения;
- экстрапирамидные нарушения, такие как непроизвольные движения рук, ног и головы.
Лечение бокового амиотрофического склероза
На сегодняшний день не разработано способов победить само заболевание, поэтому лечение сводится к двум задачами:
- Замедлить прогрессирование болезни.
- Уменьшить выраженность отдельных симптомов болезни для улучшения качества жизни.
С первой задачей справляются два препарата: рилузол и эдаравон. Рилузол — ингибитор высвобождения глутамата, одобренный для лечения БАС в 1995 году. Препарат, как правило, хорошо переносится, но обладает ограниченной эффективностью. Он продлевает жизнь примерно на 3 месяца, не улучшая качество жизни [5]. Ранее предпринимались попытки определить подгруппы пациентов, которые с большей вероятностью получат пользу от лечения рилузолом, но они были малорезультативны [6].
Препарат эдаравон является поглотителем свободных радикалов. Это внутривенное лекарственное средство для лечения БАС, которое было одобрено агентством Министерства здравоохранения США в мае 2017 года (примерно через 2 года после его одобрения для лечения БАС в Японии и Южной Корее). Использование этого препарата было оценено при разных неврологических расстройствах, включая БАС, в нескольких клинических испытаниях. Результаты были примечательными. Отмечалось замедление снижения показателей функциональной шкалы на 33 % за 6 месяцев и замедление ухудшения качества жизни людей с БАС в целом. Относительно расстройств дыхания не удалось достичь статистически значимых результатов.
Со второй задачей справляется арсенал лекарств и медицинских приборов для симптоматического лечения (например аппарат неинвазивной вентиляции лёгких при нарушении дыхательной функции) [12].

Сейчас учёные активно разрабатывают генетическую терапию при БАС, т. к. непосредственное изменение работы мутированного гена может смягчить критические этапы в развитии заболевания. Также в течение последних нескольких лет наблюдался значительный интерес к терапии на основе стволовых клеток. Современные подходы к стволовым клеткам в первую очередь предназначены для защиты выживших двигательных нейронов с помощью паракринного эффекта (процесса, в котором стволовые клетки «заставляют» окружающие клетки регенерировать). Они не предназначены для замены мёртвых моторных нейронов, но позволяют инициировать процесс роста зачатков клеток [15].
Многие пациенты используют дополнительные и альтернативные методы лечения, например витамины и пищевые добавки [7]. Некоторые добавки имеют научное обоснование (т. е. в теории должны помогать), но не имеют положительных подтверждений на практике.
Прогноз. Профилактика
Прогноз при БАС всегда неблагоприятный. В редких случаях, если подтверждена определённая мутация в гене супероксиддисмутазы-1 (D90A и некоторые другие), прогноз ещё хуже. В среднем болезнь длится 2,5-3,5 года. Лишь 7 % больных живут дольше 5 лет [3]. Длительность заболевания меньше при бульбарном дебюте БАС (прогрессирующем бульбарном параличе), при возрасте начала младше 45 лет, а также при быстром типе прогрессирования. Однако известно о двух необъяснимых случаях, когда заболевание «остановилось». Это Стивен Хокинг, который прожил с болезнью более полувека, и гитарист Джейсон Беккер, живущий с болезнью почти 30 лет. Хотя часто слышен скепсис в сторону диагноза Стивена Хокинга, так как он никогда не публиковал историю своей болезни.
Профилактика заключается в исключении или снижении влияния факторов риска: необходимо отказаться от курения; не контактировать с вредными веществами (пестицидами, гербицидами, свинцом и др.); исключить микротравмы головы.
Сообщалось, что омега-3 в сочетании с витамином Е снижает риск БАС до 60 % [11]. В связи с этим возникает вопрос, стоит ли покупать соответствующие витамины и добавки? Стоит ли доверять нутрициологам, которые объясняют все существующие болезни нехваткой того или другого витамина или микроэлемента? Сложно ответить однозначно на эти вопросы, однако существуют исследования, которые предостерегают от бездумного применения витаминов. В 2014 году были опубликованы результаты, согласно которым избыток антиоксидантов (тот самый витамин Е) увеличивает риск рака у подопытных мышей [13].
Появляются и другие исследования, опровергающие пользу «витаминотерапии» для стран, не испытывающих проблем голода и нехватки пищи среди населения. В связи с этим более целесообразно рекомендовать пациентам покупать продукты «средиземноморской диеты», т. е. употреблять в пищу естественные витамины, антиоксиданты, жирные кислоты, не опасаясь передозировки этих веществ. Жителям отдалённых регионов с материальной точки зрения иногда выгоднее покупать пищевые добавки для непродолжительного курса приёма, чем продукты средиземноморской диеты.
Медицинский эксперт статьи
Новые публикации
Болезни мотонейрона
, медицинский редактор
Последняя редакция: 19.11.2021

х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
- Код по МКБ-10
- Причины
- Симптомы
- Формы
- Диагностика
- Лечение
- К кому обратиться?
Болезни мотонейрона характеризуются неуклонно прогрессирующей дегенерацией кортико-спинальных путей, нейронов переднего рога, бульварных двигательных ядер или комбинацией этих поражений. Среди симптомов: мышечная слабость и атрофии, фасцикуляции, эмоциональная лабильность и слабость дыхательных мышц. Диагностика включает исследование скорости проведения возбуждения, ЭМГ и исключение других нарушений при нейровизуализации и в лабораторных тестах. Лечение болезни мотонейрона симптоматическое.
 [1]
[1]
Код по МКБ-10
G12.2 Болезнь двигательного неврона
Причины болезней мотонейрона
Существует несколько форм болезни мотонейрона, и их этиология часто неизвестна. Номенклатура и симптомы зависят от локализации преимущественного поражения. Среди миопатий, симулирующих клиническую картину болезни мотонейрона, поражения мышечной мембраны, сократительного аппарата и органелл миоцитов.
[2]
Симптомы болезней мотонейрона
Принято различать поражение верхнего (центрального) и нижнего (периферического) мотонейрона; иногда (например, при боковом амиотрофическом склерозе) поражаются оба нейрона.
При поражении верхнего мотонейрона (например, первичный боковой склероз) страдают нейроны на протяжении от моторной коры до ствола мозга (кортико-бульбарные тракты) или спинного мозга (кортикоспинальные тракты). Среди симптомов скованность, затрудненные и неловкие движения, сначала мышц рта, горла, потом и конечностей.
При поражениях нижнего мотонейрона страдают нейроны передних рогов спинного мозга или их эфферентные аксоны, идущие к скелетным мышцам. При бульбарном параличе поражаются только бульбарные ядра черепных двигательных нервов в стволе мозга. Обычны жалобы на слабость лицевых мышц, дисфагию и дизартрию. При поражении нейронов передних рогов, как в случае спинальной амиотрофии, обычны жалобы на слабость и атрофию, фасцикуляции (видимые мышечные мелкие подергивания) и крампи в кистях, стопах или языке. К болезням нижнего мотонейрона относят также полиомиелит и энтеровирусную инфекцию, когда поражаются нейроны передних рогов, и постполиомиелитический синдром.
Физикальное обследование помогает дифференцировать поражения верхнего и нижнего мотонейрона, а также слабость при поражениях нижнего мотонейрона от слабости при миопатиях.
Формы
[3]
Боковой амиотрофический склероз (БАС)
БАС (болезнь Лу Герига, синдром Шарко) — наиболее распространенная форма поражения мотонейрона. Заболевание начинается с асимметричных крампий, слабости и амиотрофии кистей (чаще) или стоп. Затем появляются фасцикуляции, спастичность, повышение глубоких сухожильных рефлексов, разгибательные подошвенные рефлексы, скованность движений, уменьшение массы тела, утомляемость и затруднение контроля за выражением лица и движениями языка. Среди других симптомов дисфония, дисфагия, дизартрия и поперхивание жидкой пищей. Затем появляются неуместные, непроизвольные и неконтролируемые (псевдобульбарный синдром) приступы смеха или плача. Чувствительность, сознание, когнитивная сфера, произвольные движения глаз, половая функция, а также функция сфинктеров обычно не нарушены. Смерть наступает вследствие паралича дыхательных мышц, половина больных погибают в первые 3 года от дебюта заболевания, 20 % живут 5 лет, а 10 % — 10 лет. Выживание в течение 30 лет — редкость.
[4], [5], [6], [7], [8], [9], [10]
Прогрессирующий бульбарный паралич
Нарушения функции мышц, иннервируемых черепными нервами, и кортико-бульбарных путей вызывают прогрессирующие затруднения при жевании, глотании, разговоре, появляется гнусавость голоса, снижение рвотного рефлекса, фасцикуляции и слабость мимической мускулатуры, а также языка. При поражении кортико-бульбарного пути развивается псевдобульбарный паралич с эмоциональной лабильностью. При дисфагии прогноз плохой, респираторные осложнения в результате аспирации приводят к гибели за 1-3 года.
Прогрессирующая мышечная атрофия
Во многих случаях, особенно если заболевание дебютирует в детстве, оно наследуется по аутосомно-рецессивному типу. В иных случаях оно появляется спорадически. Вообще заболевание может развиться в любом возрасте. Возможно поражение только нейронов передних рогов либо оно более выражено, чем сопутствующее поражение кортико-спинальных путей. Заболевание прогрессирует медленнее, чем другие поражения мотонейрона. Самым ранним проявлением могут быть фасцикуляции. Мышечное похудание и слабость начинаются с кистей, затем распространяются на руки, плечи и ноги. Выживаемость обычно превышает 25 лет.
Первичный боковой склероз и прогрессирующий псевдобульбарный паралич
При прогрессирующем псевдобульбарном параличе постепенно нарастает напряжение и слабость в дистальных отделах, поражая конечности, а также мышц, иннервируемых каудальными черепными нервами. Много позже могут появиться фасцикуляции и мышечная атрофия. Через несколько лет эти нарушения приводят к полной инвалидизации.
Диагностика болезней мотонейрона
Заболевание следует заподозрить при прогрессирующей генерализованной двигательной слабости, не сопровождающейся значимыми нарушениями чувствительности. Среди других неврологических заболеваний, приводящих к изолированной мышечной слабости, нарушения нервно-мышечной передачи и различные миопатии. Приобретенные причины только двигательной слабости: невоспалительные миопатии, полимиозит, дерматомиозит, поражения щитовидной железы и надпочечников, электролитные нарушения (гипокалиемия, гиперкальциемия, гипофосфатемия) и различные инфекции (например, сифилис, лаймская болезнь, гепатит С).
При поражении черепных нервов вторичная причина менее вероятна. Признаки поражения нижнего и верхнего мотонейрона, а также слабость мышц лица свидетельствуют в пользу амиотрофического бокового склероза.
Для исключения нарушений нервно-мышечной передачи и демиелинизации нервов проводят электродиагностические исследования. При поражении МН скорость проведения возбуждения обычно не нарушена вплоть до поздней стадии заболевания. Наиболее информативна игольчатая ЭМГ, демонстрирующая фибрилляции, положительные волны, фасцикуляции, а иногда и гигантские потенциалы действия двигательных единиц, даже во внешне не пораженных конечностях.
Необходимо проведение МРТ. При отсутствии клинических и ЭМГ-данных, свидетельствующих о поражении черепных нервов, назначают МРТ шейного отдела.
Для выявления потенциально излечимых заболеваний выполняют общий анализ крови, определяют уровень электролитов, креатинфосфокиназы, гормонов щитовидной железы, белка в сыворотке крови и моче, электрофорез с иммунофиксацией на моноклональные антитела, выявляют антитела к миелин-ассоциированному гликопротеину (MAG), а если есть подозрение на интоксикацию тяжелыми металлами, то исследуют их содержание в суточной моче. Следует выполнить люмбальную пункцию: повышенное содержание лейкоцитов или белка предполагает другой диагноз.
При малейших подозрениях проводят VDRL-реакцию на сифилис, определяют СОЭ, ревматоидный фактор, антитела к боррелии, ВИЧ, вирусу гепатита С, антинуклеарные антитела (ANA), антитела к нейрональным антигенам, появляющиеся в рамках паранеопластического синдрома (анти Hu). Генетическое тестирование (например, мутация гена супероксиддисмутазы) и определение ферментов (например, гексозаминидазы А) показано только в том случае, если больной заинтересован в генетическом консультировании, а результаты этих исследований никак не могут повлиять на лечение.
[11], [12], [13], [14], [15]
К кому обратиться?
Лечение болезней мотонейрона
Специфического лечения болезни мотонейрона нет. Антиглутаматный препарат рилузол по 50 мг внутрь 2 раза/день продлевает жизнь при бульбарной форме бокового амиотрофического склероза. Противостоять прогрессирующей неврологической дисфункции следует силами специалистов различного профиля. Физиотерапия помогает поддержать функцию мышц. Важно порекомендовать ортопедические фиксирующие бандажи и приспособления для ходьбы. Логопед может подобрать адекватные коммуникативные устройства. При фарингеальной слабости прием пищи представляет реальную угрозу и может потребоваться чрескожная эндоскопическая гастростомия.
При развитии респираторной слабости пульмонолог порекомендует неинвазивную дыхательную поддержку (например, двухуровневое положительное давление), трахеостомию или полную ИВЛ.
Баклофен уменьшает спастичность, квинин или фенитон могут ослабить крампи. Антихолинергические препараты (например, гликопирролат, амтриптилин, бензтропин, тригексифенидил, аппликации гиосцина, атропин) уменьшают слюнотечение. Амитриптилин и флувоксамин применяют при псевдобульбарных поражениях. В поздних стадиях этих заболеваний боль может потребовать назначения опиоидов и бензодиазепинов. Хирургическое вмешательство с целью улучшения глотания при прогрессирующем бульварном параличе малоэффективно.
Чтобы определить уровень допустимого вмешательства, лечащий врач уже в ранних стадиях заболевания болезни мотонейрона должен откровенно поговорить с пациентом, членами его семьи и теми, кто осуществляет уход. Впоследствии эти решения надо периодически пересматривать и подтверждать.

Боковой амиотрофический склероз (БАС) — идиопатическое нейродегенеративное заболевание двигательных отделов центральной нервной системы (ЦНС) человека с абсолютной летальностью. В этом обзоре авторы суммируют последние данные о происхождении заболевания, предрасположенности к нему и обсуждают вопрос о том, почему не все случаи БАС похожи друг на друга. Надо сказать, что спустя 150 лет после первого описания склероза Шарко (J-M. Charcot) прогресс в поиске ответов на данные вопросы едва ли сдвинулся с места. Авторы фокусируют внимание на том, что известно об этой болезни и в каком направлении движутся исследования, начиная с небольших шагов по увеличению продолжительности жизни, улучшению качества терапевтического ухода, прохождению клинических испытаний и компилированию эпидемиологических данных, а заканчивая комплексными мерами по предотвращению БАС и обнаружению триггеров этого заболевания.
С 90-х годов прошлого века интерес ученых и клиницистов к БАС постоянно возрастал. Прогресс в нашем понимании работы глутаматных систем, а также открытие генов, связанных с развитием наследственного БАС, значительно стимулировали исследовательский интерес: были получены данные, позволившие осознать клиническое разнообразие БАС. Переносимость болезни пациентами в данный момент зависит от нескольких факторов, включая клиническую форму, темп развития заболевания, раннее проявление респираторной недостаточности и режим питания. Увеличение продолжительности жизни пациентов с БАС, по-видимому, зависит от степени нашего понимания его патогенеза. Развитие представления о БАС позволит разработать ранние и специфические диагностические методы для выявления этого заболевания. Крайне важно сформировать терапевтическую стратегию, которая не только замедлит прогрессирование заболевания, но и позволит справиться с последствиями недостаточного питания и дыхательной недостаточности.
В настоящий момент не существует диагностического теста или биомаркера, который указывал бы на наличие БАС, и неврологи при постановке диагноза полагаются исключительно на клинические показатели. Разработка инновационных биомаркеров для объективной оценки тяжести заболевания обещает значительно усовершенствовать дизайн клинических исследований в этой области и удешевить их. К тому же, регистры населения в наши дни все чаще признаются обязательными дополнениями к клинической диагностике. Эти совместные усилия непременно приведут к лучшему пониманию патогенеза БАС, его зачастую непредсказуемых последствий и будут способствовать созданию руководств для улучшения качества ухода за такими пациентами. В этом обзоре авторы предоставляют актуальную характеристику ключевых разработок, связанных с БАС.
Проводить эпидемиологические исследования БАС затруднительно по нескольким причинам, включая сложность определения точной даты начала заболевания и потенциально длительный промежуток между появлением патологических изменений на молекулярном уровне и клиническими проявлениями болезни. Наличие продромального периода между началом заболевания и появлением у пациента первых симптомов, возможно, указывает на существование в нервной системе избыточных популяций нейронов. Ряд сложно организованных эпидемиологических исследований, в которых были задействованы обширные группы пациентов, обеспечил ученых обилием данных о различных механизмах развития заболевания. В ходе популяционных исследований было установлено, что заболеваемость БАС в Европе составляет примерно 2,6 на 100 000 человеко-лет. Несмотря на то, что проблема БАС затрагивает население всего мира, точная цифра заболеваемости до сих пор неизвестна. Заболеваемость у мужчин составляет примерно 3,0 на 100 000 человек (доверительный интервал [ДИ] 95 % в диапазоне 2,8–3,3); у женщин — 2,4 на 100 000 человек (ДИ 95 % в диапазоне 2,2–2,6). Тем не менее, частота заболеваний в случае семейного БАС у представителей обоих полов примерно одинакова. Суммарный риск развития БАС в течение жизни составляет 1:400 для женщин и 1:350 для мужчин. В возрасте 58–63 года особенно вероятно развитие спорадического БАС, 47–52 года — пик манифестации наследственного БАС. Вероятность появления БАС после 80 лет резко снижается.
Несмотря на то, что проявления БАС в различных популяциях могут показаться одинаковыми, присутствуют едва заметные различия в его клинических аспектах в зависимости от части Европы, где он наблюдается. Благодаря популяционным исследованиям получены доказательства меньшей вероятности возникновения БАС у людей смешанного происхождения, чем у людей с испанскими корнями. В исследовании населения Кубы летальность оказалась на 60 % меньше, чем в случае популяций Европы или Северной Америки. Это подтверждает предыдущие наблюдения, в ходе которых была выявлена сниженная частота возникновения БАС среди населения Северной Америки с испанскими корнями.
Примерно 5–10 % случаев БАС наследуются по генетическим законам Менделя. На момент написания этой статьи (2011 год) были открыты 13 генов и локусов, значительно влияющих на вероятность возникновения данного заболевания (при этом многие их них были открыты лишь после 2009 года). Из числа уже известных генов мутации в SOD1 (кодирует Cu2+/Zn2+-связывающую супероксиддисмутазу), TARDBP (также известный как TDP43; кодирует TAR-ДНК-связывающий белок), FUS (кодирует слияние клеток в саркоме — Fusion in Sarcoma), ANG (кодирует рибонуклеазу ангиогенин, RNase A family 5) и OPTN (кодирует синтез оптинейрина) вызывают типичную клиническую форму БАС. Мутации SOD1 индуцируют усиление функции кодируемого фермента до достижения токсического эффекта, однако его точные патофизиологические механизмы все еще остаются неясными. TDP43 и FUS (также известен как TLS-транслируемый в липосаркоме [translated in liposarcoma]) — мультифункциональные протеины, вовлеченные в экспрессию и регуляцию генов, включая транскрипцию, сплайсинг РНК, транспорт и трансляцию. FUS и TDP43 также участвуют в процессинге небольших регуляторных РНК (микроРНК) и в созревании и сплайсинге обычной РНК. Экспрессия гена ANG повышается в ответ на гипоксию, а его продукт регулирует транскрипцию РНК. OPTN — ген, определяющий развитие первичной открытоугольной глаукомы. Мутации этого гена, которые вызывают БАС, препятствуют началу активации Nf kB (nuclear factor kappa-light-chain-enhancer of activated B-cells — фактор транскрипции, контролирующий экспрессию генов иммунного ответа, апоптоза и клеточного цикла) и изменяют распределение оптинейрина в цитоплазме. Мутации в гене SOD1 вызывают 20 % наследственных форм БАС и 5 % предположительно спорадических. Мутации в TARDBP вызывают 5–10 % наследственных форм БАС, мутации в FUS — 5 %, а мутации в ANG — примерно 1 %. Оставшиеся 90 % людей, у которых был диагностирован БАС, считаются обладателями спорадического заболевания. Исследования семейного анамнеза таких пациентов указывают на некоторую связь между БАС и другими нейродегенеративными заболеваниями. Это может быть связано с наличием «генов восприимчивости», которые могут увеличивать суммарный риск деградации нервных клеток у членов одной семьи. Однако попытки упорядочить сложный генетический базис спорадического БАС при помощи идентификации этих «генов восприимчивости» не имели большого успеха. Результаты исследований генов-кандидатов позволили идентифицировать некоторые «гены восприимчивости», но относительный вклад каждого потенциально опасного гена редко превышал отношение шансов 2 к 1, а механизм увеличения риска так и остался невыясненным. Несмотря на разочаровывающие результаты нескольких недавних попыток полногеномного анализа ассоциаций, в ходе этих исследований были идентифицированы некоторые гены, предположительно ответственные за развитие спорадического БАС. Главной проблемой данных исследований явилась их низкая мощность вследствие небольших размеров образцов, к тому же полученный результат не удалось реплицировать на второй популяции. Недавнее исследование, проведенное совместно двумя группами ученых, позволило открыть два восприимчивых гена; это демонстрирует, что данные гены и пути их репликации можно идентифицировать более эффективно, если исследовательские группы станут сотрудничать между собой. Скромные достижения полногеномного анализа ассоциаций привели к переосмыслению гипотезы «одна болезнь — один вариант» в пользу гипотезы «одна болезнь — множество разных редких вариантов».
Знание различных разновидностей БАС важно для понимания возможных путей прогрессии заболевания. Идентификация специфических признаков БАС имеет важное значение не только для пациента с точки зрения прогнозирования его выживаемости, но и для проведения клинических испытаний. Основные варианты БАС включают:
- БАС с первичным поражением конечностей с признаками дегенерации верхних и нижних мотонейронов;
- первичный бульбарный БАС, представленный затруднениями речи и глотания, далее переходящий на конечности (рис. 1);
- менее распространенный первичный боковой склероз с наличием признаков поражения исключительно верхних мотонейронов;
- прогрессирующая мышечная атрофия с поражением исключительно нижних мотонейронов.
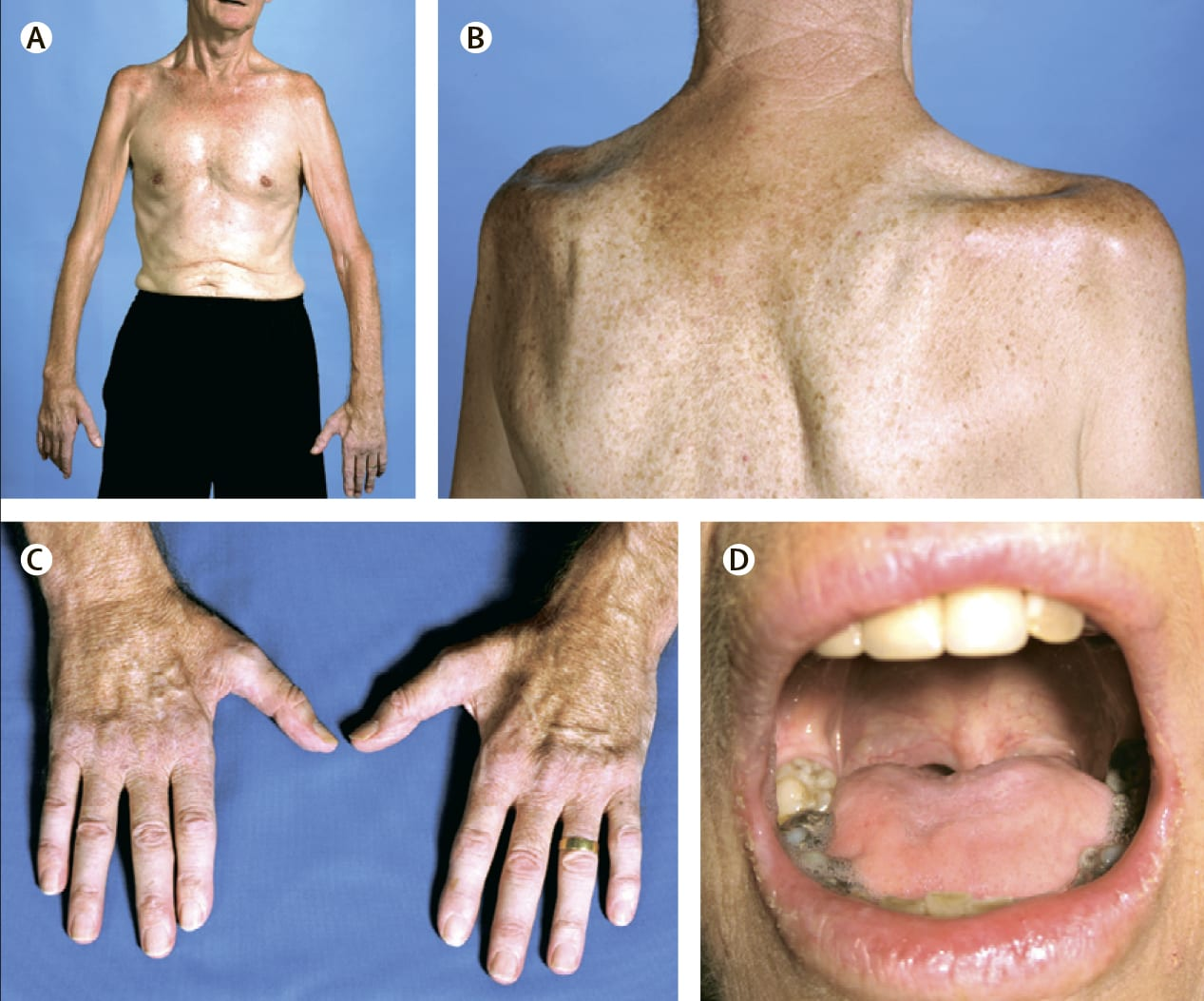
Рисунок 1 | Основные клинические характеристики БАС
Симметричная атрофия проксимальной группы мышц верхних конечностей (А) выражается в виде неспособности поднять руки против силы гравитации («человек в бочке» или «flail-arm»-вариант склероза). Стоит обратить внимание на углубления над и под лопаточной остью (Б), указывающие на атрофию надостной и подостной мышц вкупе со значительной потерей массы дельтовидной мышцы. Как следствие, плечевой сустав становится выступающим и может смещаться (С). Непропорциональная атрофия мышц возвышения большого пальца и первой тыльной межкостной мышцы (так называемая «разделенная рука») — типичный признак заболевания. Несмотря на то, что природа механизмов, которые приводят к слабости мышц кисти, пока точно не ясна, была предложена кортикомотонейронная гипотеза. Мышцы возвышения большого пальца и первая тыльная межкостная мышца получают более объемную кортикоспинальную иннервацию и, следовательно, с большей вероятностью могут быть подвержены эксайтотоксическому эффекту глутамата. (Д) Значительная атрофия мышц языка при БАС, возникшем в продолговатом мозге. Стоит обратить внимание на парез мягкого нёба при произнесении звуков. Трудности с открытием рта и дисфагия могут потребовать введения вспомогательного питания с использованием перкутанной эндоскопической гастростомии. Также в поддержку кортикомотонейронной гипотезы говорит тот факт, что у пациентов с бульбарным склерозом язык часто поражается больше, чем мышцы рта и глотки. Так же, как и мышцы возвышения большого пальца на кисти, язык получает кортикальную иннервацию обильнее, чем другие группы мышц соседних областей.
Клинически БАС проявляется в виде поражений верхних и нижних мотонейронов, которые наблюдаются в стволе мозга и во множестве участков спинного мозга. Примерно 25 % пациентов могут иметь первичную патологию продолговатого мозга, 70 % — первичную патологию, отражающуюся на конечностях, 5 % — патологию с вовлечением тела/дыхательной системы, которая впоследствии распространяется на другие области организма. Атипичные признаки могут включать потерю веса, которая является неблагоприятным прогностическим признаком, а также судороги, фасцикуляции (непроизвольные сокращения пучков мышечных волокон) без ослабления мышц, эмоциональную лабильность и фронтальную когнитивную дисфункцию долевого типа. Поражение верхних мотонейронов нарушает работу конечностей, что приводит к параличу, слабости, быстрым и резким сухожильным рефлексам. В свою очередь, признаки поражения нижних мотонейронов, связанных с конечностями, включают фасцикуляции, мышечную утомляемость и слабость. Бульбарная дисфункция верхних мотонейронов приводит к спастической дизартрии, которая характеризуется медленной, трудновыговариваемой, отрывистой речью и ринолалией. При открытии рта и смещении нижней челюсти могут возникать патологические отрывистые движения. Дисфункция нижних мотонейронов бульбарного отдела идентифицируется по утомляемости, слабости и фасцикуляции мышц языка, вялой дизартрии и последующей дисфагии. Вялая дизартрия приводит к ринолалии, вызванной слабостью мышц мягкого нёба, хрипотой и небольшим кашлем.
БАС прогрессирует безжалостно: 50 % пациентов умирают через 30 месяцев после появления первых симптомов, примерно 20 % выживают на протяжении 5–10 лет. С низкой выживаемостью связывают преклонный возраст на момент появления симптоматики, раннюю дисфункцию дыхательных мышц и манифестацию заболевания с поражения ствола мозга. Выживаемость повышают следующие независимые друг от друга факторы: болезнь, начавшаяся с нарушений в иннервации конечностей, начало заболевания в молодом возрасте и длительная задержка постановки диагноза.
Некоторые разновидности БАС предрасполагают к позитивному прогнозу. В частности, БАС «разболтанных рук» (flail-limb) (рис. 1А, 1Б) и прогрессирующая мышечная атрофия, преимущественно возникающие при поражениях нижних мотонейронов, прогрессируют медленнее, чем другие формы БАС. При изолированном бульбарном параличе, когда преобладают поражения верхних мотонейронов, а симптоматика ограничена мышцами рта и глотки, прогноз выживаемости колеблется от двух до четырех лет; эта форма чаще поражает женщин старше 65 лет. Кроме того, патология пациентов с первичным боковым склерозом прогрессирует медленнее, чем при классическом БАС. Дифференциальную диагностику первичного бокового склероза следует отложить хотя бы на четыре года с момента начала заболевания, так как возникновение поражения нижних мотонейронов может произойти даже в том случае, если первичные признаки заболевания напоминают спастический синдром.
Отличать разные клинические формы БАС важно для клинических исследований схем лечения, которые потенциально могут облегчить течение заболевания. Слабость и снижение толерантности к физической нагрузке — распространенные симптомы заболевания, которые вынуждают многих пациентов просить помощи в повседневных делах. У большинства больных БАС развивается дисфагия, что приводит к последующим недоеданию и потере веса, которые ассоциируются с плохим прогнозом. В большинстве случаев склероза развивается дыхательная недостаточность, приводя при физической нагрузке к диспноэ, ортопноэ, гиповентиляции и в результате к гиперкапнии, а также к головным болям по утрам. Смерть становится неизбежной, как только у пациента возникает диспноэ в состоянии покоя. Кроме того, прогрессирующая слабость дыхательных мышц приводит к респираторной недостаточности, часто осложняемой пневмонией.
Практически у всех пациентов с БАС и более чем у 50 % пациентов с фронтотемпоральной деменцией (ФТД) недавно идентифицировали убиквитинированные TDP43-положительные цитоплазматические включения, что вновь подогревает интерес к сходству этих прогрессирующих нейродегенеративных синдромов. Несмотря на то, что явные когнитивные симптомы и «чистая» деменция упоминались в более ранних исследованиях, они не считались характерными признаками БАС. В то же время изредка у пациентов с ФТД развивается БАС. Известно о случаях наличия в семьях обоих расстройств одновременно. Гены, которые вызывают проявление БАС и ФТД в одной семье, пока еще неизвестны, но исследования выявили наличие общего для обеих болезней локуса девятой хромосомы. Проблемы с мышлением вначале могут быть неявными, и иногда их не замечают, но при грамотной когнитивной и нейрофизиологической оценке от 20 до 50 % пациентов с БАС соответствуют критериям предположительной или подтвержденной ФТД. Среди наиболее часто встречающихся проблем: гиперфункция речи или обострение личностных качеств с когнитивным профилем, который больше всего напоминает поведенческую ФТД. В рамках клинических наблюдений проблемы с вынесениями суждений, импульсивностью и общее ухудшение способности выполнять рутинные каждодневные задачи могут вылиться в серьезные проблемы с ведением таких пациентов. Нарушение словесной коммуникации, которое ярче выражено у пациентов с псевдобульбарной формой склероза, не позволяет им сообщить о своих самых простых нуждах. Когнитивная и особенно исполнительная дисфункции могут также влиять на согласие пациента на лечение, на его способность принимать самостоятельные решения, а также потенциально осложнить этические и юридические вопросы терапии.
Схожесть этих заболеваний подтверждается воксельной МРТ-морфометрией, с помощью которой выявляются сходные структурные аномалии в ЦНС пациентов с БАС и ФТД-БАС. Может развиваться билатеральная атрофия моторной и премоторной коры, однако пациенты с ФТД-БАС обычно обладают более выраженной фронтотемпоральной атрофией, чем пациенты, имеющие только БАС. Фронтотемпоральный гиперметаболизм был выявлен у пациентов с БАС и ФТД-БАС при помощи 2-18-флуоро-2-дезокси-D-глюкозной ПЭТ. Данная фронтотемпоральная атрофия, по-видимому, связана с потерей нейронов и кортикальным глиозом, обнаруживаемыми на аутопсии. Так же, как и для большинства пациентов со спорадическим склерозом, у 50 % пациентов с ФТД присутствуют TDP43-положительные внутринейронные включения. Недавно у пациентов с убиквитин-положительной, TDP43-отрицательной ФТД и пациентов с семейным БАС, вызванным мутациями в FUS, были обнаружены FUS-положительные включения. Это вновь акцентирует внимание на взаимосвязи БАС и ФТД.
Патофизиологические механизмы, лежащие в основе развития БАС, видимо, запускаются множеством разнообразных факторов (рис. 2). Появляется все больше и больше доказательств роли комплексного взаимодействия генетических и молекулярных механизмов в развитии этого заболевания. БАС может проявляться как порок развития опорно-двигательного аппарата в зрелом возрасте. В частности, согласно контрольным исследованиям шведских ученых, с повышенным риском развития БАС были сопряжены слишком раннее или позднее материнство, а также наличие у пациентов младших родных братьев и сестер. К тому же, развитие двигательной системы человека может потенциально быть нарушено в детстве при повышенной предрасположенности к инфекциям; как правило, это случается в семьях с маленькими детьми. Также были предложены различные факторы среды, влияющие на развитие склероза, включая интенсивную физическую нагрузку в течение жизни и активную военную службу. В ретроспективном исследовании футболистов профессиональных итальянских лиг стандартизированная смертность оказалась увеличена из-за развития БАС, особенно в виде рано дебютировавших форм. По неизвестным причинам футболисты, которые играли более пяти лет, в особенности на позиции атакующего полузащитника, имели наибольший риск развития БАС. Несколько случаев склероза также было зафиксировано среди футболистов, выступающих в Англии.
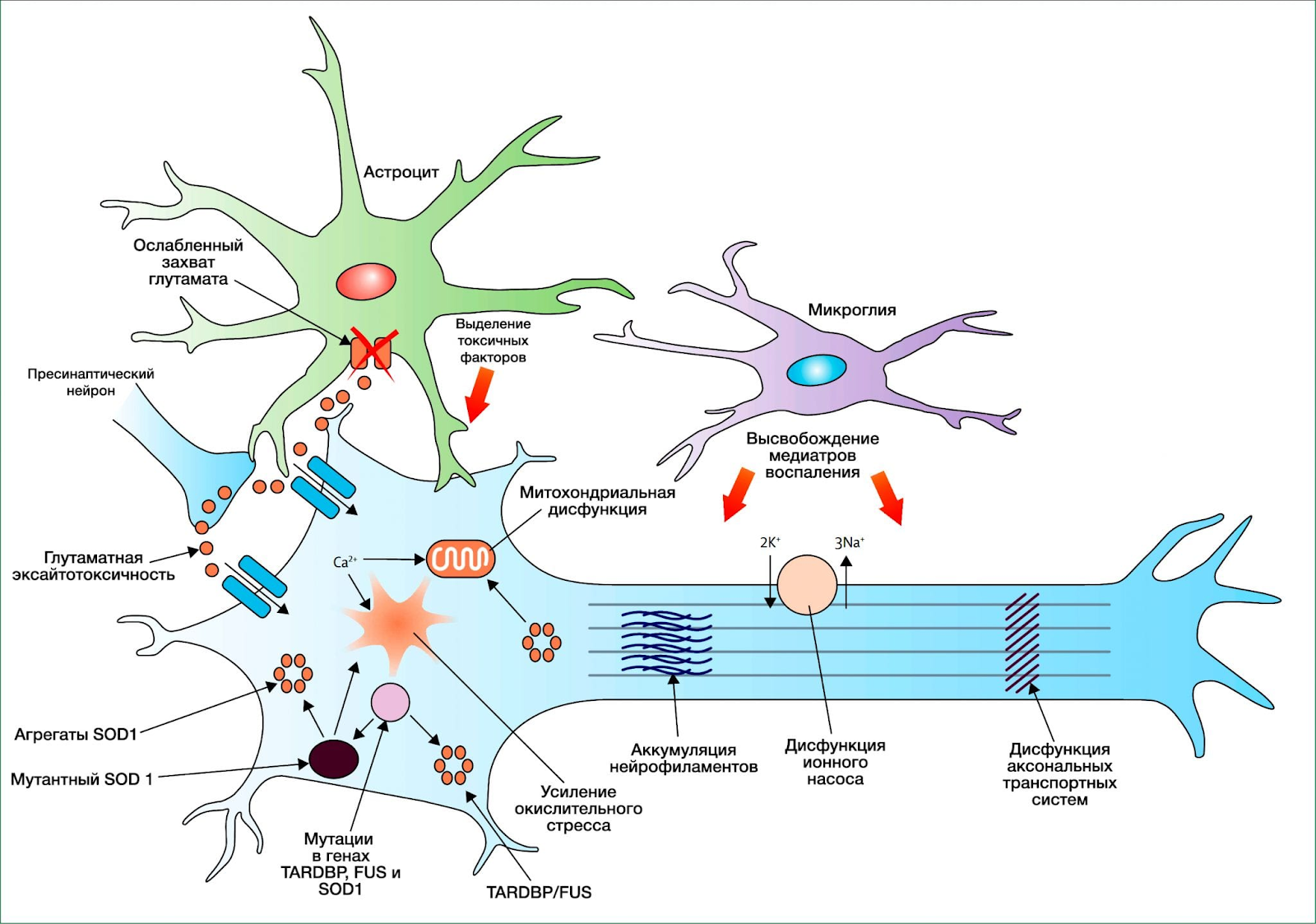
Рисунок 2 | Клеточные и молекулярные механизмы, обуславливающие нейродегенерацию при БАС
Механизмы, вызывающие дегенерацию нейронов при БАС, мультифункциональны и работают на молекулярном и генетическом уровнях. В частности, дегенерация нервных клеток при БАС может возникнуть из-за целого комплекса нарушений: глутаматной эксайтотоксичности; синтеза свободных радикалов; агрегации цитоплазматических белков; деятельности SOD1 ферментов, сопровождаемой митохондриальной дисфункцией и нарушением процесса аксонального транспорта вследствие аккумуляции агрегатов внутриклеточных нейрофиламентов. Мутации в TARDBP и FUS выражаются в формировании внутриклеточных агрегатов, которые оказывают вредоносное воздействие на нейроны. Активация микроглии приводит к выработке провоспалительных цитокинов, что приводит к интоксикации организма. В конце концов, деградация мотонейронов может происходить вследствие активации кальций-зависимых ферментативных путей.
Курение в определенных дозах может оказывать эффект на последующее развитие БАС. Влияние нейротоксинов, включая β-метил-амино-L-аланин, на организм человека связывают с возникновением эпидемии заболевания БАС-Паркинсона на острове Гуам. Эта нейротоксичная аминокислота концентрировалась в мозге пациентов с болезнью БАС-Паркинсона и включалась в рацион Чаморро (коренные жители о. Гуам) из-за употребления аборигенами в пищу летучих мышей. Эти мыши, блюда из которых являются деликатесами для Чаморро, питаются семенами орхидей (Cymbidium), содержащими высокие концентрации β-метил-амино-L-аланина.
Не было установлено четкой связи между мутациями в SOD1 и ранней гибелью мотонейронов. Текущее представление объясняет возникновение генетических мутаций чрезмерной токсичной функцией фермента SOD1 и синтезом свободных радикалов, что в конечном счете приводит к повреждению и гибели клетки. К тому же, мутации SOD1 индуцируют конформационную нестабильность и неправильное сворачивание белка SOD1, способствуя формированию внутриклеточных включений, что ингибирует нормальную активность протеасомы, нарушая тем самым системы аксонального транспорта и другие жизненно важные функции клетки.
В схему патогенеза БАС также вовлечена эксайтотоксичность глутамата. Глутамат — основной возбуждающий нейромедиатор в ЦНС. Глутамат связывается с ионотропными N-метил-D-аспартатными (NMDA) рецепторами и рецепторами α-амино-3-гидрокси-5-метил-4-изоксазоолепропионовой кислоты (AMPA) на постсинаптической мембране. Чрезмерная активация данных рецепторов глутаматом может спровоцировать нейродегенерацию вследствие активации кальций-зависимых ферментативных путей. Индуцированный глутаматом токсический эффект может также привести к синтезу свободных радикалов, что может вызвать нейродегенерацию посредством разрушения внутриклеточных органелл и влияния на синтез провоспалительных медиаторов. Механизм, при помощи которого токсический эффект глутамата вызывает деградацию мотонейронов у людей, остается неясным (сноска 1, рис. 3). Был предложен так называемый механизм «смерти наперед». Согласно ему, верхние мотонейроны вызывают антероградную деградацию нижних мотонейронов при оказании глутаматом своего токсического эффекта.
Сноска 1 | Дилемма БАС: где именно начинается заболевание?
Несмотря на начальное наблюдение Шарко одновременных патологических изменений верхних и нижних мотонейронов при БАС, вопрос точной локализации начала заболевания остается открытым. Нахождение ответа на него могло бы прояснить патофизиологию БАС; это имеет большое диагностическое и терапевтическое значение. Гипотеза «смерти наперед» предполагает, что БАС — синдром, затрагивающий в основном кортикальные мотонейроны, которые напрямую соединяются с клетками передних рогов спинного мозга, вызывая их антероградную дегенерацию посредством эксайтотоксичного действия глутамата. Ниже представлены некоторые факты, подтверждающие гипотезу «смерти наперед».
Результаты исследований путем транскраниальной магнитной стимуляции, которые показывают, что кортикальное гипервозбуждение является ранним признаком наличия у пациентов спорадического БАС и предшествует первичным клиническим симптомам наследственной формы заболевания.
Клинические наблюдения: (1) мотонейроны без прямой связи с кортикальными нейронами, принадлежащие ядрам глазодвигательного (III) и отводящего (IV) нервов и ядру Онуфа, обычно сохраняются при БАС; (2) отсутствие случаев БАС среди животных в природных условиях объясняется недостаточным количеством у последних связей между кортикальными мотонейронами и клетками передних рогов; (3) чистые формы БАС нижних мотонейронов редки, в то время как бессимптомные поражения верхних мотонейронов без исключения обнаруживаются с помощью транскраниальной магнитной стимуляции.
Гипотеза «обратной смерти» предполагает, что БАС начинает свое развитие с мышечных клеток или нервно-мышечных синапсов. В частности, при этом наблюдается дефицит двигательного нейротрофического гормона. В норме этот гормон выделяется постсинаптическими клетками и ретроградно транспортируется по аксону пресинаптической клетки к ее телу, где оказывает определенный эффект. В число доказательств гипотезы «обратной смерти» входят следующие наблюдения:
• началу деградации мотонейронов предшествует денервация;
• денервация опосредуется аккумуляцией мутировавшего белка SOD1 в шванновских клетках.В противовес гипотезам «смерти наперед» и «обратной смерти» существуют предположения некоторых исследователей о том, что процессы деградации верхних и нижних мотонейронов происходят независимо друг от друга.
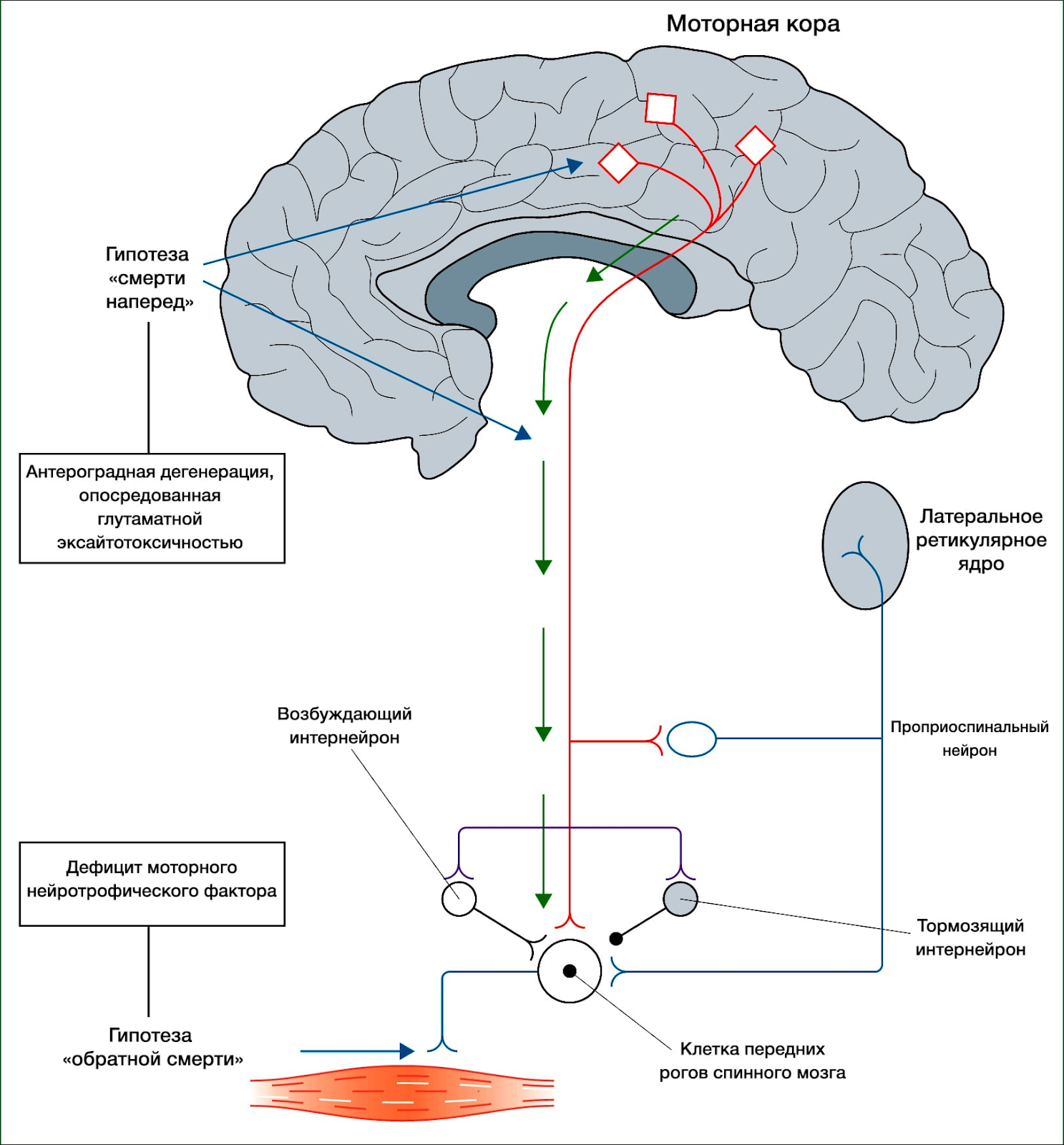
Рисунок 3 | Гипотезы «смерти наперед» и «обратной смерти»
Кроме вышеуказанной токсичности, в развитие патогенеза БАС вовлечены структурные аномалии митохондрий, дисфункция калий-натриевой помпы, аутофагоцитоз и нарушения аксонального транспорта. Не только нейроны, но и такие клетки, как астроциты и микроглиоциты, также могут вызывать нейродегенерацию путем недостаточной секреции нейротрофических факторов, секреции нейротоксичных медиаторов и модуляции экспрессии рецепторов глутамата (все это известно как нейродегенерация по вне-нейронным причинам). Следующим по важности в патофизиологии склероза является TDP43, который был определен как основной компонент убиквитинированных цитоплазматических белковых агрегатов у всех пациентов со спорадическим БАС, но расположенный вне ядра (в нормальных нейронах он находится именно там). Несмотря на то, что вопрос того, являются ли данные агрегаты причиной нейродегенерация при БАС, остается открытым, мутации в TARDBP были обнаружены всего лишь в 3 % случаях наследственной формы склероза и у 1,5 % пациентов со спорадическим БАС, позволяя предположить, что агрегаты TDP43 играют ключевую роль в инициации БАС. Доказательство патогенности мутации TARDBP было представлено, когда мутации, идентифицированные в крайне защищенных участках ДНК, не вызвали БАС в контрольных группах. Учитывая, что TDP43 связывается и с ДНК, и с РНК, мутации в TARDBP могли бы привести к нарушению регуляции процессинга РНК. Данную теорию поддерживает обнаружение мутаций гена FUS на 16-й хромосоме, которые связаны с наследственными формами БАС. Агрегаты FUS не были явно определены у пациентов с патологическими изменениями в TDP43 или SOD1, что указывает на наличие нового пути возникновения болезни. Несмотря на то, что обнаружение мутации в генах TARDBP и FUS, которая вызывала склероз, было наибольшим шагом в понимании его патогенеза, другие факторы также требуют внимания. Приводят ли мутации в данных ДНК/РНК-связывающих белках к гиперфункции, ведущей к токсическому эффекту и дисфункции? Чем именно вызывается нейротоксичность? Дефектом в свертывании белков, который нарушает систему внутриклеточной передачи сигналов? Или она опосредована уничтожением жизненно важных белков и генетического материала агрегатами TDP43 и FUS? Какова связь между недавно обнаруженными патофизиологическими механизмами и белками TDP43 и FUS? Ученым только предстоит найти ответы на эти вопросы.
В отсутствие диагностического теста на БАС клиницисты в большинстве случаев полагаются на идентификацию признаков дегенерации верхних и нижних мотонейронов в одной области организма с последующим наблюдением динамики распространения заболевания. Критерии El Escorial, опубликованные в 1997 году, используют совокупность признаков поражения верхних и нижних мотонейронов для проведения точной диагностики на надлежащем уровне. Исследователи в клинических испытаниях систематизировали пациентов с возможным или точно диагностированным БАС согласно критериям El Escorial, отмечая их универсальность; несмотря на это, использование данных диагностических признаков в качестве критериев для классификации пациентов может в некоторых случаях оказаться не самым лучшим вариантом. Дело в том, что данные критерии могут быть недостаточно показательными, особенно на ранних стадиях развития БАС, когда наиболее сильны эффекты терапевтических воздействий на пациентов. Благодаря критике, критерии модифицировали для упрощения ранней диагностики и более точной постановки диагноза, что важно в клинической практике.
Часто постановке четкого диагноза предшествует длительный период времени, отчасти из-за того, что первые симптомы обнаруживаются в среднем лишь через 14 месяцев после дебюта. Необычные клинические симптомы, не вызывающие у врача должных подозрений, неправильная интерпретация результатов нейрофизиологических и нейрорадиологических исследований — вот некоторые распространенные причины неясностей в процессе диагностики. К сожалению, задержка постановки диагноза может привести к применению ошибочных терапевтических стратегий, позднему назначению подходящей терапии, а также проблемам при взаимодействии с пациентом с точки зрения его психологического фона. Процесс диагностики БАС тяжело переносится пациентом и членами его семьи, следовательно, его необходимо строго контролировать. При бесчувственном оглашении диагноза пациенту и его родственникам эмоциональная нагрузка на них может оказаться чрезмерной; нерешительность врача при постановке диагноза в атипичных случаях может существенно осложнить процесс принятия пациентом своего заболевания и прогноза. Четкое следование расписанию визитов к врачу после постановки диагноза помогает получить ответы на вопросы, которые не были обсуждены во время первичной консультации, а также снабдит пациента ценной информацией о службах поддержки, которые хорошо организованы в большинстве развитых стран.
При дифференциальной диагностике следует помнить о том, что существует несколько расстройств, напоминающих БАС; для нахождения различий между ними обычно используют структурную визуализацию, нейрофизиологические и лабораторные исследования (сноска 2). В случаях наличия синдромов поражения исключительно нижних мотонейронов крайне важно провести генетическое тестирование на болезнь Кеннеди, сцепленную с Х-хромосомой бульбарную атрофию и спинальную мышечную атрофию. Образцы мышечной биопсии могут иметь диагностическую ценность для исключения нестандартных миопатий, таких как прогрессирующая миоклонус-эпилепсия (болезнь Лафора), или для подтверждения наличия БАС путем обнаружения атрофии волокон различных типов (рис. 4А).
Сноска 2 | Дифференциальная диагностика БАС и исследования, необходимые для его подтверждения
Расстройства мотонейронов:
• спинальная мышечная атрофия (анализ делеции гена SMN);
• сцепленная с Х-хромосомой спинобульбарная мышечная атрофия (болезнь Кеннеди; повышенное количество триплетов ЦАГ в ДНК из крови);
• полиомиелит или постполиомиелитный синдром (история болезни, электронейромиография [ЭНМГ]);
• дефицит гексозаминидазы А (тестирование ферментов лейкоцитов).Расстройства двигательных нервов:
• мультифокальная двигательная нейропатия (ЭНМГ, антитела к ганглиозиду GM1);
• хроническая воспалительная демиелинизирующая нейропатия (ЭНМГ, люмбальная пункция);
• синдром спастической фасцикуляции (ЭНМГ);
• нейромиотония (антитела к потенциал-зависимым калиевым каналам);
• наследственная предрасположенность к спастическому парапарезу (тест на генетические мутации);
• наследственная двигательная нейропатия с пирамидальными характеристиками;
• радикулоплексопатия (ЭНМГ, МРТ);
• паранеопластический синдром (сывороточные маркеры, визуализация, образец биопсии костного мозга);
• отравление тяжелыми металлами (анализ мочи или крови);
• множественные мононевриты (ЭНМГ, скрининг сосудов, серологический анализ).Расстройства нервно-мышечных соединений:
• миастения (антитела к рецепторам ацетилхолина, антитела к мышечно-специфицированной тирозинкиназе, повторная стимуляция нервных окончаний при ЭНМГ, электромиография одиночных волокон);
• миастенический синдром Ламберта-Итона (повторная стимуляция нервных окончаний при ЭНМГ).Структурные поражения ЦНС и спинного мозга:
• сирингомиелия или сирингобульбия (МРТ);
• спинная сухотка (серологический анализ на сифилис);
• рассеянный склероз (МРТ, олигоклональные цепочки иммуноглобулинов, тесты на рассеянный склероз);
• мономелическая спинальная мышечная атрофия (болезнь Хираяма; электромиография, МРТ);
• болезнь Лайма (серологическое исследование на болезнь Лайма);
• человеческий Т-лимфотропный вирус I типа (анализ на ВИЧ).Миопатия:
• миозит с включениями (электромиография, креатинфосфокиназа [КФК], образец биопсии мышцы);
• полиомиелит (электромиография, КФК, образец биопсии мышцы, аутоиммунный скрининг);
• дерматомиозит (электромиография, КФК, образцы биопсий кожи и мышц);
• прогрессирующая миоклонус-эпилепсия (Болезнь Лафора) (исследования проводимости нервов, электромиография, образец биопсии мышцы или нерва).Эндокринная система:
• тиреотоксикоз (тест на функционирование щитовидной железы, электромиография, образец биопсии мышцы);
• гиперпаратиреоидизм (концентрация ионов кальция и исследование околощитовидных желез);
• подострая комбинированная дегенерация спинного мозга (болезнь Лихтгейма) (измерение концентрации витамина В12);
• целиакия (серологическая диагностика, образец биопсии кишечника).
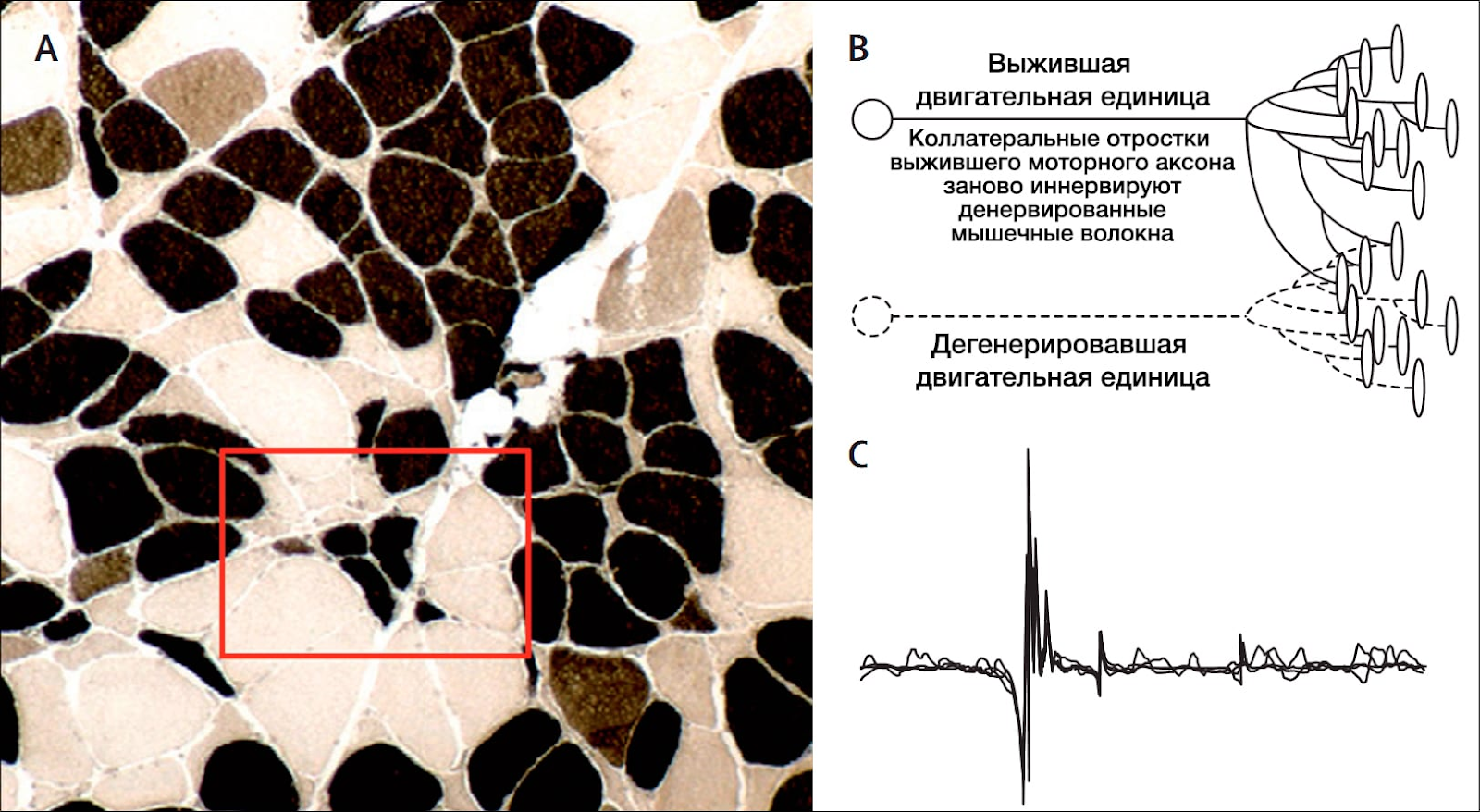
Рисунок 4 | Результаты исследований пациента с БАС
(А) Образец биопсии левой мышцы vastus lateralis пациента с БАС, окрашенный АТФазой при pH 9,4. На образце биопсии выделены сгруппированные атрофированные волокна I и II типов (волокна смешанного типа, заключенные в красный прямоугольник). (В) Патофизиология дегенерации и реиннервации двигательных единиц; с наложением (С) друг на друга 10 записей, которые демонстрируют типичные крупные полифазные нестабильные (комплексные) двигательные единицы, отмечаемые при стабилизировавшемся БАС (длительность амплитуды записи 50 миллисекунд) с запаздывающими составляющими компонентами, указывающими на реиннервацию.
Рутинные нейрофизиологические исследования пациентов с БАС включают в себя изучение синапсов, электромиографию и транскраниальную магнитную стимуляцию (реже). Исследование нервных соединений необходимо для исключения расстройств, которые напоминают БАС, в особенности демиелинизирующих двигательных нейропатий. На ранних стадиях развития склероза проводимость двигательных нервов не нарушена, но по мере прогрессирования болезни общая амплитуда потенциала действия в мышце снижается, демонстрируя денервацию. Проводимость чувствительных нервов у пациентов со склерозом обычно не нарушена, что отличает БАС от демиелинизирующих нейропатий. Заметные аномалии, обнаруженные в ходе изучения проводимости чувствительных нервов, должны вызывать подозрения клиницистов на предмет наличия альтернативных диагнозов. У пациентов с преимущественным поражением нижних мотонейронов возможно наличие излечимых расстройств, таких как мультифокальная двигательная нейропатия. Эту возможность нужно учитывать при наличии блокады проводимости хотя бы двух двигательных нервов вне типичных мест проявления БАС.
Электромиография в дополнение к исследованиям проводимости нервов также полезна для идентификации дисфункции нижних мотонейронов (рис. 4Б). Помимо дисфункции нижних мотонейронов, она также демонстрирует фибриллярные потенциалы, положительные остроконечные волны и хронические нейрогенетические изменения (рис. 4С). Данные электромиографические аномалии недавно были включены в обновленные критерии диагностики БАС El Escorial, дополняя признаки поражения нижних мотонейронов. Фибриллярные потенциалы и остроконечные волны могут наблюдаться в мышцах, которые внешне выглядят нормально. Следовательно, электромиография может помочь в ранней диагностике склероза, указывая на наличие субклинического вовлечения деградации нижних мотонейронов. Сохранившиеся двигательные единицы могут спонтанно деполяризовываться в виде потенциалов фасцикуляции, которые клинически проявляются в виде непроизвольных мышечных подергиваний — типичного признака БАС. В частности, высокоспецифичными для этого заболевания являются подергивания мышц языка.
Наличие фасцикуляций в отсутствие других электромиографических феноменов следует интерпретировать с осторожностью, ведь это может указывать на менее серьезные расстройства, особенно на «доброкачественный» синдром спастической фасцикуляции. С другой стороны, недавно исправленные и приведенные к консенсусу руководства (известные как критерии острова Аваджи) приравнивают фасцикуляции к фибриллярным потенциалам у пациентов с подозрением на БАС. К тому же, фасцикуляции в случае БАС являются комплексными («злокачественными»): они указывают на реиннервацию и наличие хронических нейрогенных изменений (рис. 4С).
В настоящее время активно обсуждается то, насколько успешно врачи диагностируют БАС, используя клинические критерии и данные лабораторных исследований. На этом акцентируется внимание из-за статистики по БАС в Шотландии, где доля ложноположительных результатов составила 8 %. Другая информация из популяционных исследований указывает на схожие ложноположительные цифры; количество же ложноотрицательных результатов достигло 44 %. Основными причинами пересмотра ложноположительных результатов были отсутствие прогрессирования симптоматики по данным исследований, возникновение атипичных признаков, а также противоречивые результаты сопутствующих нейрофизиологических и нейрорадиологических исследований. Наиболее часто по ошибке диагностируют как БАС такие заболевания, как мультифокальная двигательная нейропатия и болезнь Кеннеди.
Наибольший вклад нейровизуализации в диагностику БАС на данный момент внесла способность МРТ исключать альтернативные причины его возникновения. Однако данная сфера активно развивается, и результаты мультимодальной нейровизуализации подтвердили тот факт, что склероз является мультисистемным церебральным нейродегенеративным расстройством. Ключевые открытия в области изыскания биомаркеров заболевания, сделанные при помощи нейровизуализации, описываются в сноске 3.
Сноска 3 | Ключевые открытия в нейровизуализации БАС
Гиперинтенсивность кортикоспинального пути на МРТ. Гиперинтенсивность кортикоспинальных трактов, видимая на МРТ, может быть заметна при БАС, однако она не является специфичным признаком данной болезни (рис. 5А).
Церебральная атрофия, выявленная при помощи МРТ. Воксель-зависимая морфометрия (техника «картирования» мозга, определяющая разницу в объеме его структур) позволяет измерить количество серого и белого вещества с целью обнаружения церебральной атрофии, сопряженной с когнитивным расстройством у пациентов с БАС. При этом зоны максимальной атрофии различаются у пациентов с врожденным и спорадическим БАС. Пациенты с врожденным заболеванием обладают большей продолжительностью жизни. 3D-технологии изучения мозга при помощи МРТ также могут помочь в обнаружении фокальных аномалий (рис. 6).
Магнитно-резонансная спектроскопия. Измерение количества содержащих протоны метаболитов, таких как N-ацетиласпартат (количество рассчитывается относительно отношения к (фосфо)креатину или холину), стало маркером нейронедостаточности. У пациентов с БАС отношение N-ацетиласпартата к креатину по сравнению с контрольной группой было снижено в зонах первичной моторной коры. Магнитно-резонансная спектроскопия оказалась очень чувствительной именно при обнаружении дисфункции нижних мотонейронов, что позволило отличать пациентов с прогрессирующей мышечной атрофией от пациентов с БАС.
Диффузионная тензорная визуализация (диффузионная МРТ). Диффузионную МРТ можно использовать для идентификации степени диффузии воды, показатели которой считаются константными (анизотропными) внутри нетронутых болезнью проводящих путей и более диффузными (изотропными) в путях со сниженной интегративностью. Количественные единицы измерения, такие как частичная анизотропность и основной коэффициент диффузии, являются мощными суррогатными маркерами патологических изменений в нервной ткани. Внутренняя связь между нервными путями может быть картирована при помощи техники связующей трактографии (рис. 5Б). Использование диффузионной тензорной визуализации также может продемонстрировать сниженную частичную анизотропность кортикоспинальных путей у пациентов с БАС.
Функциональные исследования. Результаты позитронной эмиссионной томографии (ПЭТ) 2-18-флюоро-2-дезокси-D-глюкозой и H2O указали на широко распространенные экстрамоторные изменения у пациентов, страдающих склерозом с фронтальной недостаточностью. Последняя была сопряжена с нейропсихологическими повреждениями, демонстрируя проявляющийся в клинике перекрест БАС и ФТД. Неинвазивные исследования активации мозга с помощью функциональной МРТ используют разницу резонаторных свойств оксигемоглобина и дезоксигемоглобина (функциональная МРТ, зависимая от оксигенации крови). При помощи анализа BOLD (функционального МРТ) всего мозга в покое можно идентифицировать функционально связанные участки мозга. Результаты исследований пациентов с БАС показали и «стандартный режим» работы мозга, и изменения в ней при активации сенсомоторной сети. Эта техника имеет потенциал в дальнейшем более схематично представлять экстрамоторные церебральные патологические изменения у пациентов.
Молекулярная визуализация. Лиганд-рецепторная ПЭТ может быть использована для исследований молекулярных механизмов болезни. Результаты C-флумазениловой ПЭТ продемонстрировали сниженные ингибирующие ГАМКергические кортикальные эффекты у людей с БАС, учитывая гипотезу кортикальной гипервозбудимости как фундаментальный аспект его патогенеза. Использование рецепторного к бензодиазепину лиганда C-PK11195 при ПЭТ выявило обширную активацию микроглии при БАС, вместе с которой были обнаружены биомаркеры воспаления в спинномозговой жидкости. Установленное в лобнотеменных зонах снижение способности лиганда C-WAY100635 связывать рецептор 5-HT1A у пациентов с БАС и результаты исследований нейропатологических рецепторов, которые показывают схожие изменения при ФТД, позволяют предположить, что наличие серотонинергических механизмов оправдывает дальнейшие исследования патогенеза склероза. В конечном счете, парамагнетические свойства небольших частиц оксида железа, которые могут быть использованы как интравенозные контрастные агенты, могут указывать на начало эпохи молекулярной МРТ, что потенциально позволит не только понять механизмы воспаления, но и использовать стволовые клетки в терапевтических целях.
Определение пресимптоматических маркеров заболевания. Нечеткое представление группы риска в случае спорадического БАС препятствует нахождению раннего (пресимптоматического) биомаркера. Результаты исследований с помощью диффузной спектральной томографии пациентов пресимптоматической группы с ярко выраженной мутацией в гене SOD1 демонстрируют изменения в задней ножке внутренней капсулы, не отмеченные в контрольных группах здоровых людей. Возможно, это одно из первых изменений, которое можно обнаружить у пациентов. Респираторная функция и питание пациентов с БАС являются важными проблемами, ведь респираторная недостаточность является основной причиной смерти. Были разработаны экспертные рекомендации для решения ключевых вопросов, ассоциированных с БАС, среди них проблемы с дыхательной системой, питанием и уходом за пациентами. При следовании рекомендациям необходимо делать акцент на наиболее благоприятных потенциальных вариантах развития болезни.
Средством специфической нейропротективной терапии пациентов с БАС является рилузол — ингибитор высвобождения глутамата (сноска 4). В двух крупных независимых друг от друга исследованиях этот препарат увеличил продолжительность жизни пациентов на 3–6 месяцев. Его преимущества лучше проявлялись при назначении в специализированных многопрофильных клиниках для больных БАС, чем в других учреждениях. Рилузол приносил наибольшую пользу пациентам с умеренными функциональными повреждениями нервной системы.
Симптоматическая терапия остается краеугольным камнем в ведении пациентов с БАС (сноска 5). Для некоторых пациентов эти методы не только смягчают симптоматику, но и повышают продолжительность и качество жизни. Оптимальный уход за пациентами с БАС предоставляется в условиях многопрофильной среды, где физиотерапевты, реабилитологи, логопеды, пульмонологи, гастроэнтерологи и социальные работники сотрудничают между собой и обеспечивают пациентам симптоматическое лечение. Подобные многопрофильные подходы снизили риск смерти в течение 5 лет болезни на 45 %, что делает их потенциальным средством повышения выживаемости. По сравнению с пациентами, которые содержались в клинике общей неврологии, находящиеся в специализированной клинике жили более качественной жизнью, что, возможно, связано с более эффективным использованием ресурсов, которое начинает приносить пациентам пользу уже после первого визита к врачу.
Сноска 4 | Полемика вокруг БАС — клинические исследования
Несмотря на то, что клинические исследования БАС проводятся с 1980-х годов, рилузол неизменно остается единственным лекарством с доказанной эффективностью против данного расстройства. Неудовлетворительные результаты клинических исследований БАС, которые так и не привели к каким-либо конкретным результатам, были объяснены наличием проблем в структуре исследований на доклиническом и клиническом уровнях.
Проблемы на доклиническом уровне
Неудовлетворительные результаты имитации заболевания на мышах. До недавнего времени SOD1-модель на мышах являлась стандартом тестирования нейропротекторов, используемых при БАС. Однако, так как мутации SOD1 составляют примерно 2 % случаев склероза, имитация на мышах не может быть сопоставима со спорадической болезнью человека. К тому же, в процессе имитации происходят стереотипные изменения, которые начинаются со слабости задних конечностей у мышей. Модель на последних с мутацией гена, кодирующего TDP43, — потенциальное продвижение в развитии терапии БАС. Оно предоставляет ученым обновленную и, возможно, более релевантную платформу для изучения новейших программ терапии.
Неправильно выбранное время назначения лекарств и проблемы с дозировкой. Некоторые исследователи изучили влияние пресимптоматического применения лекарств в начале развития заболевания. Несмотря на то, что это может внести свой вклад в наше понимание субклинических процессов, которые предшествуют дегенерации мотонейронов, по-видимому, оно имеет небольшое отношение к собственно лечению спорадического БАС. Многие доклинические исследования также проверили действие ультравысоких доз лекарств, которые, возможно, достигнут такой концентрации в плазме, что не будут переноситься пациентами. Некоторые исследователи рекомендуют не назначать пациентам максимально переносимую дозу в погоне за наилучшим эффектом.
Проблемы на клиническом уровне
Суть исследования. Существует растущая потребность в более эффективном скрининге фармакологических средств на протяжении второй фазы испытаний, так как после данного периода принимается решение: продолжить подтвержденное тестирование (т. е. начать третью фазу испытаний) либо отказаться от лекарства как от неэффективного. В связи с этим трудно провести грань между второй и третьей фазой исследования БАС, ведь предоставление предварительного доказательства эффективности лекарства во второй фазе затруднительно. Главной причиной этого является отсутствие эффективного биомаркера. Следовательно, следует либо использовать эффективные статистические стратегии для минимизирования продолжительности исследования и размера выборок, либо увеличить шанс успеха на третьей фазе испытаний (что приведет к повышению количества ложноположительных результатов), либо проводить третью фазу испытаний, используя методику второй фазы (что приведет к повышению количества ложноотрицательных результатов). Последний подход, возможно, один из тех, что сейчас используются редко в силу недавнего прогресса в дизайне статистических исследований. Ученые также продолжают работать с третьей фазой клинических испытаний, даже в отсутствие предварительных доказательств их эффективности препаратов у человека.
Выбор первичной конечной точки. Изменения первичной конечной точки производятся в зависимости от успешности испытания. Выбор правильной первичной конечной точки при клинических испытаниях БАС является дискутабельным вопросом. Преобладают испытания, которые используют функциональные линейки и силовые показатели как первичные конечные точки. Испытания, целью которых было изыскание средств, увеличивающих выживаемость, до недавнего времени были редкими. Смысл и преимущества последних включают меньший размер выборок, меньшую продолжительность испытаний и клинически значимый эффект от лечения. Тем не менее, измерение числа выживших может проводиться только при доказанной эффективности лечения, ведь с начала первичных симптомов потери мотонейронов слишком велики. Примером средства, увеличивающего выживаемость, является рилузол. Он оказывает небольшие, но значимые эффекты, которые можно обнаружить уже при работе с пробами, размер которых меньшего необходимого для повышения выживаемости.
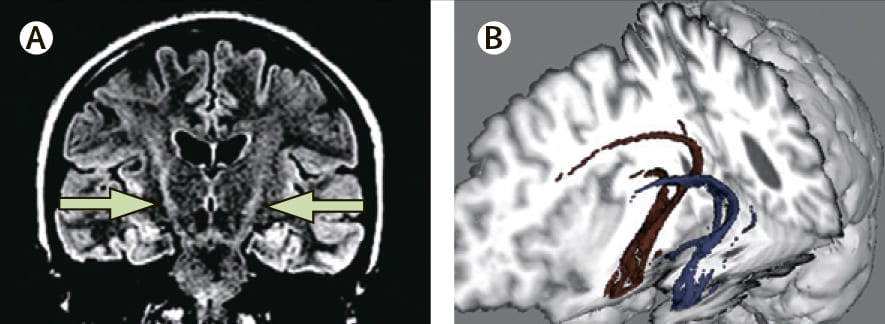
Рисунок 5 | Стандартное и экспериментальное МРТ-исследования пациентов с БАС
(А) Режим Т2-FLAIR (режим Т2 с подавлением сигнала от свободной жидкости — прим. ред.) демонстрирует гиперинтенсивный сигнал от кортикоспинальных путей у пациента с БАС на коронарном срезе (указано стрелками), но данный признак не является точным и специфичным для диагностики БАС в отсутствие других более очевидных клинических симптомов. (Б) Диффузионная тензорная трактография — техника МРТ, с помощью которой можно изучать вовлеченность экстрамоторных и моторных нейронных путей в патогенез БАС (верхний косой наружный срез мозга, рассматриваемый слева). У данного пациента с необычным фенотипом склероза, включающим выраженную афазию, при реконструкции белых проекционных волокон височной доли обнаружено меньшее количество волокон с левой стороны (синий цвет), чем с правой (красный цвет).
Рисунок 6 | 3D-МРТ головного мозга пациента с первичным боковым склерозом, вид сверху
Стрелки указывают на значительно расширенные прецентральные борозды с относительной атрофией прилежащих долей, особенно моторных. Макроскопическая атрофия, видимая в данном случае, является редкой для пациентов с типичным БАС, но чаще заметна у пациентов с первичным боковым склерозом, у которых заболевание манифестирует преимущественно поражением верхних мотонейронов. Этот рисунок демонстрирует позднюю стадию кортикомотонейронного патологического процесса, который считается неотъемлемым для любого типа БАС.
Респираторная недостаточность указывает на одновременную деградацию нейронов центральных дыхательных центров и мотонейронов, отдающих аксоны в диафрагмальный нерв. В клинической картине пациентов с БАС часто присутствуют респираторные нарушения. Ночная гипоксия и ассоциированные с ней симптомы летаргии, потеря концентрации, головные боли по утрам и неудовлетворительный сон — последствия дисфункции ЦНС. Слабость диафрагмы и постепенное снижение жизненной емкости легких (ЖЕЛ) можно диагностировать методом спирометрии. Показатели силы мышц, совершающих вдох, такие как максимальное давление на вдохе и давление при вдыхании носом, являются более точными маркерами дыхательной дисфункции, чем ЖЕЛ; в случае пациентов со стабильной слабостью лицевой мускулатуры (т. е. тех, кто не может держать рот в широко открытом состоянии) они могут быть более информативными.
Сноска 5 | Симптоматический уход при БАС
Слабость и утомляемость:
• биопротезирование (протезы стопы, шейные воротники);
• физиотерапия;
• адаптивные элементы поддержки (ходунки, инвалидное кресло).Дисфагия:
• наблюдение у речевого терапевта и диетолога;
• техники безопасного глотания и модифицированная диета;
• введение гастростомической трубки.Диспноэ и слабый кашель:
• вентиляционная поддержка;
• морфин или бензодиазепины;
• физиотерапия области груди;
• отсасывание мокроты;
• помощь при откашливании.Боль (мышечная и костная; судороги, фасцикуляции и паралич, боль при надавливании на кожу, вызванная ее неподвижностью):
• физиотерапия, нестероидные противовоспалительные препараты (НПВП);
• миорелаксанты (баклофен, ботулинический токсин);
• противосудорожные препараты (например, габапентин);
• помощь при изменении положения тела и уход за областями, которые подвергаются повышенному давлению, во избежание образования пролежней;
• опиоидные препараты;
• снижающие давление тела подушки и матрасы.Дизартрия:
• наблюдение логопеда;
• вспомогательные средства общения;
• предоставление членам семьи и специалистам по уходу за пациентом всей необходимой информации.Когнитивные изменения (дисфункция лобной доли или деменция):
• объяснение симптоматики специалистам по уходу за пациентами и членам их семей;
• терапия антидепрессантами.Гиперсаливация:
• антихолинергические антидепрессанты (амитриптилин);
• антихолинергические препараты (гликопиррония бромид);
• инъекции ботулотоксина;
• облучение слюнных желез;
• продукты для ухода за полостью рта;
• отсасывание слюны.Уплотнение слюны:
• натуральные лекарства (на основе папайи);
• достаточное потребление воды;
• соляные небулайзеры; небулизированный N-ацетилцистеин;
• отсасывание слюны из ротовой полости;
• уход за ротовой полостью.Эмоциональная лабильность:
• предоставление необходимой информации членам семьи и специалистам по уходу за пациентом;
• амитриптилин;
• бензодиазепины;
• декстрометорфана гидробромид/хинидин сульфат.Депрессия и беспокойство:
• консультация специалиста;
• бензодиазепины;
• антидепрессанты.Расстройства сна:
• решение исходной проблемы;
• осмотр дыхательной системы, неинвазивная вентиляция;
• бензодиазепины, трициклические антидепрессанты.Запоры:
• модификация диеты (повышение потребления воды и грубой волокнистой пищи);
• использование в приготовлении пищи отрубей и волокнистых ингредиентов;
• регулярный прием оральных слабительных (мовикол или суппозитории).
Несмотря на то, что проведение полисомнографии может быть оптимальным подходом для идентификации приступов ночной гипоксии, для пациентов с БАС, как правило, лучше подходит ночная пульсоксиметрия. Неинвазивная вентиляция улучшает качество жизни пациентов и повышает выживаемость. К данному методу рекомендуется прибегать при наличии следующих признаков: симптомы слабости дыхательной мускулатуры (диспноэ и ортопноэ), низкая концентрация кислорода в крови согласно ночной оксиметрии, повышенное парциальное давление углекислого газа (PCO2) менее 65 мм Hg и сниженная ЖЕЛ (менее 80 %), давление вдыхания носом менее 40 см H2O. Пациенты с травмой продолговатого мозга и гиперсаливацией могут не перенести неинвазивную вентиляцию, поэтому в их случае важен соответствующий уход за дыхательными путями и выведение избыточного секрета. У пациентов с БАС, которые не переносят этот тип вентиляции, или в случае, когда его недостаточно в силу развивающейся слабости респираторных мышц, одним из вариантов является инвазивная вентиляция трахеостомией. Несмотря на то, что этот метод продлевает жизнь, данный подход редко используется в большинстве стран из-за последующих практических проблем, большой стоимости и сильной потере качества жизни. Важно заметить, что в рамках симптоматической терапии значительное облегчение пациентам, страдающим диспноэ в покое, предоставляет подкожное введение морфина.
Недоедание — ключевой фактор в определении прогноза заболевания. Развитие недоедания при БАС имеет множество причин, включая сниженное количество употребляемой пищи вследствие дисфагии и гиперметаболизма. Примерно 50–60 % пациентов с БАС пребывают в состоянии гиперметаболизма, которое персистирует на протяжении заболевания и зависит от возраста, пола и массы тела без учета жировой ткани. Повышение в показателях метаболизма, измеренное подсчетом высвобожденной в покое энергии, ассоциировано со сниженной выживаемостью. Несмотря на то, что механизмы, провоцирующие гиперметаболическое состояние, остаются неясными, известно, что дисфункция митохондрий миоцитов вносит вклад в этот аспект патогенеза БАС. Перкутанное введение гастростомической трубки обеспечивает достаточное потребление калорий и жидкости и должно производиться пациентам со значительной потерей веса, в том числе и в отсутствие дисфагии. Целесообразность проведения гастростомии следует обсудить на ранних этапах заболевания, поскольку смертность повышается, когда жизненная емкость не превышает 50 % от нормы. Внимание ко многим симптомам, которые могут развиться на протяжении заболевания, важно для повышения качества жизни пациентов с БАС (сноска 5). Терминальная стадия заболевания может сопровождаться беспокойством, тревогой, болью и диспноэ, поэтому пациентам необходимо предоставить хорошо организованный мультидисциплинарный уход. В конечном счете пациенты могут также искать альтернативные стратегии лечения (сноска 6), часто стоящие больших денег и не обладающие доказанной эффективностью в борьбе с болезнью.
Сноска 6 | Дебаты вокруг БАС — альтернативные и недокументированные способы лечения
Тот факт, что пациенты все чаще хотят экспериментировать с научно не доказанными методами терапии, неудивителен, учитывая то, что диагноз БАС является терминальным. Популярные альтернативные и недокументированные стратегии лечения включают: инсулиноподобный фактор роста 1 (ILGF1), карбонат лития, миноциклин и терапию стволовыми клетками. Пациентам следует соблюдать осторожность, обращаясь к таким вариантам лечения. Согласно некоторым клиническим исследованиям, многие из них могут ускорить процессы ослабления мышц и негативно сказаться на выживаемости. Недавно был создан интернет-ресурс ALSUntangled для информирования сообщества пациентов с БАС об альтернативных и недокументированных способах лечения заболевания. ALSUntangled позволяет пациентам и клиницистам обмениваться информацией о подобных способах лечения склероза. Пациенты с БАС поддерживают обмен новыми гипотетическими альтернативами и методами лечения, так как цель данного проекта — именно суммировать и распространить информацию о цене, научных и этических основах этих методов, а также о потенциальных преимуществах и рисках каждого нового варианта лечения.
«Давайте же несмотря ни на что продолжим поиски! Это, вне сомнений, лучший способ открыть новое, и, возможно, именно благодаря этим нашим попыткам вердикт, который мы вынесем пациенту завтра, уже не будет таким, каким мы его выносим сегодня» — Жан-Мари Шарко, 1889
В отличие от незначительного прогресса в течение прошлого века, недавние продвижения в понимании генетики, клинических проявлений и патофизиологии БАС поддерживают реалистичную надежду, что в будущем появятся абсолютно новые подходы к его лечению.
Нашли опечатку? Выделите фрагмент и нажмите Ctrl+Enter.