From Wikipedia, the free encyclopedia
| Schwann cell | |
|---|---|

The PNS has satellite cells and Schwann cells. |
|
| Identifiers | |
| MeSH | D012583 |
| FMA | 62121 |
| Anatomical terms of neuroanatomy
[edit on Wikidata] |
Schwann cells or neurolemmocytes (named after German physiologist Theodor Schwann) are the principal glia of the peripheral nervous system (PNS). Glial cells function to support neurons and in the PNS, also include satellite cells, olfactory ensheathing cells, enteric glia and glia that reside at sensory nerve endings, such as the Pacinian corpuscle. The two types of Schwann cells are myelinating and nonmyelinating.[1] Myelinating Schwann cells wrap around axons of motor and sensory neurons to form the myelin sheath.
The Schwann cell promoter is present in the downstream region of the human dystrophin gene that gives shortened transcript that are again synthesized in a tissue-specific manner.
During the development of the PNS, the regulatory mechanisms of myelination are controlled by feedforward interaction of specific genes, influencing transcriptional cascades and shaping the morphology of the myelinated nerve fibers.[2]
Schwann cells are involved in many important aspects of peripheral nerve biology—the conduction of nervous impulses along axons, nerve development and regeneration, trophic support for neurons, production of the nerve extracellular matrix, modulation of neuromuscular synaptic activity, and presentation of antigens to T-lymphocytes.
Charcot–Marie–Tooth disease, Guillain–Barré syndrome (acute inflammatory demyelinating polyradiculopathy type), schwannomatosis, chronic inflammatory demyelinating polyneuropathy, and leprosy are all neuropathies involving Schwann cells.
Structure[edit]
| Schwann cells wrapped around an axon |
|---|
|
Dendrite Soma Axon Nucleus Node of Axon terminal Schwann cell Myelin sheath |
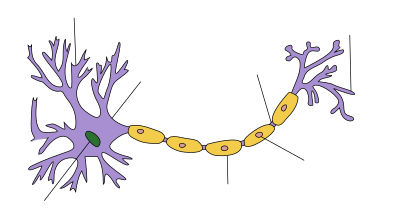
Schwann cells are a variety of glial cells that keep peripheral nerve fibres (both myelinated and unmyelinated) alive. In myelinated axons, Schwann cells form the myelin sheath. The sheath is not continuous. Individual myelinating Schwann cells cover about 1 mm of an axon[3]—equating to about 1000 Schwann cells along a 1-m length of the axon. The gaps between adjacent Schwann cells are called nodes of Ranvier.
9-O-Acetyl GD3 ganglioside is an acetylated glycolipid which is found in the cell membranes of many types of vertebrate cells. During peripheral nerve regeneration, 9-O-acetyl GD3 is expressed by Schwann cells.[4]
Function[edit]
The vertebrate`s nervous system relies on the myelin sheath for insulation and as a method of decreasing membrane capacitance in the axon. The action potential jumps from node to node, in a process called saltatory conduction, which can increase conduction velocity up to 10 times, without an increase in axonal diameter. In this sense, Schwann cells are the PNS’s analogues of the central nervous system’s oligodendrocytes. However, unlike oligodendrocytes, each myelinating Schwann cell provides insulation to only one axon (see image). This arrangement permits saltatory conduction of action potentials with repropagation at the nodes of Ranvier. In this way, myelination greatly increases speed of conduction and saves energy.[5]
Nonmyelinating Schwann cells are involved in maintenance of axons and are crucial for neuronal survival. Some group around smaller axons (External image here) and form Remak bundles.
Myelinating Schwann cells begin to form the myelin sheath in mammals during fetal development and work by spiraling around the axon, sometimes with as many as 100 revolutions. A well-developed Schwann cell is shaped like a rolled-up sheet of paper, with layers of myelin between each coil. The inner layers of the wrapping, which are predominantly membrane material, form the myelin sheath, while the outermost layer of nucleated cytoplasm forms the neurilemma. Only a small volume of residual cytoplasm allows communication between the inner and outer layers. This is seen histologically as the Schmidt-Lantermann incisure.
Regeneration[edit]
Schwann cells are known for their roles in supporting nerve regeneration.[6] Nerves in the PNS consist of many axons myelinated by Schwann cells. If damage occurs to a nerve, the Schwann cells aid in digestion of its axons (phagocytosis). Following this process, the Schwann cells can guide regeneration by forming a type of tunnel that leads toward the target neurons. This tunnel is known as band of Büngner, a guidance track for the regenerating axons, which behaves like an endoneural tube. The stump of the damaged axon is able to sprout, and those sprouts that grow through the Schwann-cell «tunnel» do so at the rate around 1 mm/day in good conditions. The rate of regeneration decreases with time. Successful axons can, therefore, reconnect with the muscles or organs they previously controlled with the help of Schwann cells, but specificity is not maintained and errors are frequent, especially when long distances are involved.[7] Because of their ability to impact regeneration of axons, Schwann cells have been connected to preferential motor reinnervation, as well.
If Schwann cells are prevented from associating with axons, the axons die. Regenerating axons will not reach any target unless Schwann cells are there to support them and guide them. They have been shown to be in advance of the growth cones.
Schwann cells are essential for the maintenance of healthy axons. They produce a variety of factors, including neurotrophins, and also transfer essential molecules across to axons.

A Schwann cell in culture.
Genetics[edit]
Schwann cell formation[edit]
Sox10[edit]
SOX10 is a transcription factor active during embryonic development and abundant evidence indicates that it is essential for the generation of glial lineages from trunk crest cells.[8][9] When SOX10 is inactivated in mice, satellite glia and Schwann cell precursors fail to develop, though neurons are generated normally without issue.[8] In the absence of SOX10, neural crest cells survive and are free to generate neurons, but glial specification is blocked.[9] SOX10 might influence early glial precursors to respond to neuregulin 1[8] (see below).
Neuregulin 1[edit]
Neuregulin 1 (NRG1) acts in a number of ways to both promote the formation and ensure the survival of immature Schwann cells.[10] During embryonic development, NRG1 inhibits the formation of neurons from neural crest cells, instead contributing to neural crest cells being led down a path to gliogenesis. NRG1 signaling is not, however, required for glial differentiation from the neural crest.[11]
NRG1 plays important roles in the development of neural crest derivatives. It is required for neural crest cells to migrate past the site of dorsal root ganglia to find the ventral regions of sympathetic gangliogenesis.[12] It is also an essential axon-derived survival factor and a mitogen for Schwann cell precursors.[13] It is found in the dorsal root ganglion and motor neurons at the point in time that Schwann cell precursors begin to populate spinal nerves and therefore influences Schwann cell survival.[11] In embryonic nerves, the transmembrane III isoform likely is the primary variant of NRG1 responsible for survival signals. In mice that lack the transmembrane III isoform, Schwann cell precursors are eventually eliminated from spinal nerves.[14]
Formation of myelin sheath[edit]
P0[edit]
Myelin protein zero (P0) is a cell-adhesion molecule belonging to the immunoglobulin superfamily and is the major component of peripheral myelin, constituting over 50% of the total protein in the sheath.[15][16] P0 has been shown to be essential for the formation of compact myelin, as P0 null mutant (P0-) mice showed severely aberrant peripheral myelination.[17] Although myelination of large caliber axons was initiated in P0- mice, the resulting myelin layers were very thin and poorly compacted. Unexpectedly, P0- mice also showed degeneration of both axons and their surround myelin sheaths, suggesting that P0 plays a role in maintaining the structural integrity of both myelin formation and the axon with which it’s associated. P0- mice developed behavioral deficits around 2 weeks of age when mice began to show signs of slight trembling. Gross incoordination also arose as the animals developed, while trembling became more severe and some older mice developed convulsing behaviors. Despite the array of impaired motor behavior, no paralysis was observed in these animals. P0 is also an important gene expressed early within the Schwann cell lineage, expressed in Schwann cell precursors after differentiating from migrating neural crest cells within the developing embryo.[18]
Krox-20[edit]
Several important transcription factors are also expressed and involved at various stages in development changing the features on the Schwann cells from an immature to mature state. One indispensable transcription factor expressed during the myelination process is Krox-20. It is a general zinc-finger transcription factor and is expressed in the rhombomeres 3 and 5.
Krox-20 is considered one of the master regulators of PNS myelination and is important in driving transcription of specific structural proteins in the myelin. It has been shown to control a set of genes responsible for interfering with this feature in the axon changing it from a pro-myelinating to myelinating state.[19] In this way, in Krox-20 double knock out mice, it has been recorded that hindbrain segmentation is affected as well as myelination of Schwann cell associated axons. Indeed, in these mice, the Schwann cells are not able to perform their myelination properly as they only wrap their cytoplasmic processes one and half turn around the axon and despite the fact that they still express the early myelin marker, late myelin gene products are absent. In addition, recent studies have also proven the importance of this transcription factor in maintaining the myelination phenotype (and requires the co-expression of Sox 10) as its inactivation leads to dedifferentiation of the Schwann cells.[2]
Clinical significance[edit]
Charcot–Marie–Tooth disease (CMT), Guillain–Barré syndrome (GBS, acute inflammatory demyelinating polyradiculopathy type), schwannomatosis, and chronic inflammatory demyelinating polyneuropathy (CIDP), leprosy, and Zika Virus are all neuropathies involving Schwann cells.[20]
Transplantation[edit]
A number of experimental studies since 2001 have implanted Schwann cells in an attempt to induce remyelination in multiple sclerosis-afflicted patients.[21] In the past two decades, many studies have demonstrated positive results and potential for Schwann cell transplantation as a therapy for spinal cord injury, both in aiding regrowth and myelination of damaged CNS axons.[22] Schwann cell transplants in combination with other therapies such as Chondroitinase ABC have also been shown to be effective in functional recovery from spinal cord injury.[23]
See also[edit]
- Electrophysiology
- Hodgkin–Huxley model
- Mesaxon
- Neurotransmission
- Olfactory ensheathing cell
- Schwannoma
- List of human cell types derived from the germ layers
References[edit]
- ^ Bhatheja, K; Field, J (2006). «Schwann cells: origins and role in axonal maintenance and regeneration». The International Journal of Biochemistry & Cell Biology. 38 (12): 1995–9. doi:10.1016/j.biocel.2006.05.007. PMID 16807057.
- ^ a b Topilko, Piotr; Schneider-Maunoury, Sylvie; Levi, Giovanni; Baron-Van Evercooren, Anne; Chennoufi, Amina Ben Younes; Seitanidou, Tania; Babinet, Charles; Charnay, Patrick (1994-10-27). «Krox-20 controls myelination in the peripheral nervous system». Nature. 371 (6500): 796–799. Bibcode:1994Natur.371..796T. doi:10.1038/371796a0. PMID 7935840. S2CID 4333028.
- ^ Tortora, Gerard J. (2017). Principles of Anatomy and Physiology (15th ed.). USA: Wiley. p. 412. ISBN 978-1-119-32064-7.
- ^ Túlio Ribeiro-Resende, Victor; Lopes, Michelle (2010). «Involvement of 9-O-Acetyl GD3 Ganglioside in Mycobacterium leprae Infection of Schwann Cells». J. Biol. Chem. 285 (44): 34086–34096. doi:10.1074/jbc.M110.147272. PMC 2962507. PMID 20739294.
- ^ Kalat, James W. Biological Psychology, 9th ed. USA: Thompson Learning, 2007.[page needed]
- ^ Bhatheja, Kanav; Field, Jeffrey (2006). «Schwann cells: Origins and role in axonal maintenance and regeneration». The International Journal of Biochemistry & Cell Biology. 38 (12): 1995–9. doi:10.1016/j.biocel.2006.05.007. PMID 16807057.
- ^ Carlson, Neil R. Physiology of Behavior, 9th ed. USA: Pearson Education, Inc., 2007.[page needed]
- ^ a b c Britisch, S.; et al. (2001). «The transcription factor Sox10 is a key regulator of peripheral glial development». Genes Dev. 15 (1): 66–78. doi:10.1101/gad.186601. PMC 312607. PMID 11156606.
- ^ a b Paratore, C., Goerich, D. E., Suter, U., Wegner, M. & Sommer, L. (2001). «Survival and glial fate acquisition of neural crest cells are regulated by an interplay between the transcription factor Sox10 and extrinsic combinatorial signalling». Development. 128 (20): 3949–3961. doi:10.1242/dev.128.20.3949. PMID 11641219.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ Shah, N. M.; et al. (1994). «Glial growth factor restricts mammalian neural crest stem cells to glial fate». Cell. 77 (3): 349–360. doi:10.1016/0092-8674(94)90150-3. PMID 7910115. S2CID 20297598.
- ^ a b Jessen, K. R. & Misky, R. (2005). «The origin and development of glial cells in peripheral nerves». Nature Reviews Neuroscience. 6 (9): 671–682. doi:10.1038/nrn1746. PMID 16136171. S2CID 7540462.
- ^ Britisch, S.; et al. (1998). «The ErbB2 and ErbB3 receptors and their ligand, neuregulin-1 are essential for development of the sympathetic nervous system». Genes Dev. 12 (12): 1825–1836. doi:10.1101/gad.12.12.1825. PMC 316903. PMID 9637684.
- ^ Dong, Z.; et al. (1995). «NDF is a neuron-glia signal and regulates survival, proliferation, and maturation of rat Schwann cell precursors». Neuron. 15 (3): 585–596. doi:10.1016/0896-6273(95)90147-7. PMID 7546738. S2CID 15332720.
- ^ Wolpowitz, D.; et al. (2000). «Cysteine-rich domain isoforms of the neuregulin-1 gene are required for maintenance of peripheral synapses». Neuron. 25 (1): 79–91. doi:10.1016/s0896-6273(00)80873-9. PMID 10707974. S2CID 16187922.
- ^ Greenfield, S.; Brostoff, S.; Eylar, E. H.; Morell, P. (1973). «Protein composition of myelin of the peripheral nervous system». Journal of Neurochemistry. 20 (4): 1207–1216. doi:10.1111/j.1471-4159.1973.tb00089.x. PMID 4697881. S2CID 30385476.
- ^ Lemke, G. (1988). «Unwrapping the genes of myelin». Neuron. 1 (7): 535–543. doi:10.1016/0896-6273(88)90103-1. PMID 2483101. S2CID 27086229.
- ^ Geise, K.; Martini, R.; Lemke, G; Soriano, P.; Schachner, M. (1992). «Mouse P0 Gene Disruption Leads to Hypomyelination, Abnormal Expression of Recognition Molecules, and Degeneration of Myelin and Axons». Cell. 71 (4): 565–576. doi:10.1016/0092-8674(92)90591-y. PMID 1384988. S2CID 41878912.
- ^ Jessen, K.; Mirsky, R. (2005). «The origin and development of glial cells in peripheral nerves». Nature Reviews Neuroscience. 6 (9): 671–682. doi:10.1038/nrn1746. PMID 16136171. S2CID 7540462.
- ^ Salzer, James (2015). «Schwann cell myelination». Cold Spring Harbor Perspectives in Biology. 7 (8): a020529. doi:10.1101/cshperspect.a020529. PMC 4526746. PMID 26054742.
- ^ Dhiman, Gaurav; Abraham, R.; Griffin, D. (2019). «Human Schwann cells are susceptible to infection with Zika and yellow fever viruses, but not dengue virus». Scientific Reports. 9 (1): 9951. Bibcode:2019NatSR…9.9951D. doi:10.1038/s41598-019-46389-0. PMC 6616448. PMID 31289325.
- ^ «First surgical transplant attempted to repair myelin». Inside MS. 2001. Archived from the original on 2007-03-11.
- ^ Oudega, Martin; Xu, Xiao-Ming (2006). «Schwann Cell Transplantation for Repair of the Adult Spinal Cord». Journal of Neurotrauma. 23 (3–4): 453–67. doi:10.1089/neu.2006.23.453. PMID 16629629.
- ^ Fouad, Karim; Lisa Schnell; Mary B. Bunge; Martin E. Schwab; Thomas Liebscher; Damien D. Pearse (2 February 2005). «Combining Schwann Cell Bridges and Olfactory-Ensheathing Glia Grafts with Chondroitinase Promotes Locomotor Recovery after Complete Transection of the Spinal Cord». The Journal of Neuroscience. 25 (5): 1169–78. doi:10.1523/JNEUROSCI.3562-04.2005. PMC 6725952. PMID 15689553.
External links[edit]
- Diagram at clc.uc.edu
- Histology image: 21301loa – Histology Learning System at Boston University—»Ultrastructure of the Cell: myelinated axon and Schwann cell»
- Cell Centered Database—Schwann cell
ВВЕДЕНИЕ
Актуальность исследований морфофункциональных особенностей шванновских клеток (SCs)
объясняется двумя причинами. Во-первых, в общебиологическом плане невозможно переоценить
значение этих миелинобразующих клеток и самого процесса миелинизации для развития
нервной системы. Во-вторых, необходимость глубоких фундаментальных знаний о закономерностях
развития и механизмах функционирования SCs связана с разрабатываемыми в настоящее
время способами стимуляции регенерации поврежденных нервных проводников.
Важное значение шванновских клеток, как клеток, осуществляющих миелинизацию нервных
волокон, обусловлено высокой значимостью возникновения миелина в эволюции [1]. С его возникновением упростилась синхронизация мышечных сокращений, нервная система
стала более компактной, появилась возможность более быстрой обработки сложной информации
[2, 3]. Salzer и Zalc рассматривают миелинизацию аксонов как возникшую в эволюции адаптивную
реакцию, способствующую развитию быстрой проводимости [4].
Одной из важнейших функций шванновских клеток является разделение (изоляция) соседних
нервных волокон.
Вторая причина, обусловливающая важность изучения шванновских клеток, связана с необходимостью
понимания молекулярно-клеточных механизмов восстановления поврежденных нервов. Изучению
строения и регенерации периферических нервных проводников посвящено множество работ
как отечественных, так и зарубежных авторов. Начало таким исследованиям было положено
в XIX веке английским нейрофизиологом Августом Валлером (1816–1870) [5]. Комплекс процессов, которые происходят в дистальном конце нервного ствола после
травмы нерва, был назван его именем – валлеровская дегенерация. Впоследствии многие
исследователи, используя нейроморфологические, гистохимические, иммуногистохимические
методы и электронную микроскопию, изучали клеточные взаимоотношения в поврежденном
нерве. В классических исследованиях зарубежных и отечественных нейроморфологов показано,
что основными структурными элементами, которые участвуют в дегенеративных и репаративных
процессах в поврежденном нерве, являются моторные, вегетативные и чувствительные нейроны,
отростки которых и составляют периферический нерв, шванновские клетки, макрофаги,
клетки кровеносных сосудов и соединительнотканные элементы оболочек нерва [5–8].
Особый интерес к проблеме, касающейся молекулярных механизмов клеточных взаимоотношений
структурных элементов периферических нервов в норме и после повреждения, возник в
последние десятилетия. Это связано с разработкой клеточных и генных технологий, которые,
как показано во многих работах и обобщено в нескольких обзорах [9–16], могут способствовать регенерации периферических нервных проводников после повреждения.
Актуальность таких исследований связана с тем, что применяемые в микрохирургической
практике способы лечения поврежденных нервов не всегда приводят к их полному функциональному
восстановлению. Одной из причин является недостаток фундаментальных нейробиологических
знаний, касающихся молекулярных механизмов регенерации нерва. Также нет полной ясности,
каковы механизмы влияния пересаженных клеток (в том числе генетически модифицированных
клеток со сверхэкспрессией ростовых факторов) на репаративные процессы в поврежденном
нерве.
Учитывая важную эволюционную роль шванновских клеток, а также их значение для регенерации
поврежденных нервных проводников, представляется важным обобщить имеющиеся в настоящее
время данные о происхождении этих клеток, их морфофункциональных особенностях, развитии
и дифференцировке, что и явилось целью настоящего обзора.
ШВАННОВСКИЕ КЛЕТКИ: МИЕЛИНИЗИРУЮЩИЕ
И НЕМИЕЛИНИЗИРУЮЩИЕ
В середине XIX века немецкий физиолог Теодор Шванн впервые описал клетки, “обернутые
вокруг аксонов периферической нервной системы” [17]. Эти клетки были названы его именем [18]. Следует отметить, что в ряде работ применяется еще один термин, обозначающий шванновские
клетки, – “леммоциты”, введенный шведским невропатологом Nils Ragnar Eugene Antoni
(1887–1968), изучавшим опухоли, характерные для оболочек нерва [19]. В трудах S. Ramon y Cajal глиальные элементы, описываемые автором в поврежденном
нерве, называются “клетками Шванна” и “леммобластами” [6]. Термины “леммоцит” и “леммобласт” и сейчас используются отдельными авторами [8, 20]. По мнению Ноздрачева и Чумасова [8], леммобласты являются одной из стадий развития шванновских клеток и отличаются от
более ранних предшественников способностью образовывать базальную пластинку. Следует
отметить, что в современной отечественной гистологической номенклатере в качестве
синонима “шванновской клетки” предложен термин “нейролеммоцит” [21].
Шванновские клетки (SCs) делятся на две группы: миелинизирующие и немиелинизирующие
клетки. И те, и другие играют ключевую роль в поддержании трофики и регенерации аксонов
нейронов в ПНС. Большинство периферических нервных проводников позвоночных животных
миелинизированы. Немиелинизированными являются аксоны малого калибра. Следует отметить,
что и в ЦНС, где в качестве миелинизирующих клеток выступают олигодендроциты, аксоны
некоторых нейронов остаются немиелинизованными, например, тонкие аксоны клеток-зерен
мозжечка, поднимающиеся в молекулярный слой коры мозжечка и ветвящиеся в нем [22].
Вопрос о том, к какой группе будут относиться шванновские клетки: к миелинизирующим
или немиелинизирующим, определяется в эмбриогенезе на стадии незрелых шванновских
клеток [23]. В настоящее время многие сигнальные пути, регулирующие развитие SCs и процесс миелинизации,
изучены [1]. Считается, что вопрос о том, будет ли аксон миелинизироваться, зависит от типа
аксона, с которым соседствуют предшественники, а также от микроокружения (белков экстрацеллюлярного
матрикса, главным образом, ламинина). Основная роль отводится нейрегулину 1 третьего
типа (NRG1-III). Аксонами большого диаметра вырабатывается высокий уровень NRG1-III,
что способствует дифференцировке предшественников в миелинизирующие шванновские клетки.
При этом одна клетка миелинизирует одно волокно. Для аксонов малого диаметра характерен
низкий уровень секреции нейрегулина, это приводит к тому, что клетки-предшественники
дифференцируются в направлении немиелинизирующих шванновских клеток [23–26]. В то же время NRG1-III необходим для дифференцировки как миелинизирующих, так и
немиелинизирующих SCs [27]. Таким образом, в дополнение к уже изученным сигнальным путям в будущем предстоит
выяснить, какие другие внешние сигналы управляют процессами развития SCs и миелинизации.
Неизвестно, имеются ли негативные сигналы на немиелинизированных нервных волокнах,
которые препятствуют миелинизации [27]. Еще предстоит выяснить, каково влияние не только аксональных сигналов, но и влияние
других типов клеток (эндотелия, фибробластов) на дифференцировку SCs в онтогенезе
[27].
Немиелинизирующие шванновские клетки имеют ряд отличительных морфологических особенностей
по сравнению с миелинизирующими. Во-первых, они располагаются вдоль аксонов на более
близком расстоянии друг от друга, во-вторых, они, как правило, находятся в контакте
более чем с одним аксоном [27, 28]. Такие клетки окружают несколько тонких (диаметром менее 1 мкм) аксонов (без формирования
миелиновой оболочки), образуя структуру, называемую пучок Ремака (“Remak bundle”).
В ПНС имеется несколько классов немиелинизированных нервных волокон: ноцицепторные
С-волокна, постганглионарные симпатические и парасимпатические волокна и терминальные
окончания двигательных нервов в нервно-мышечных синапсах [27, 29]. Немиелинизирующие шванновские клетки входят в состав телец Пачини и чувствительных
телец Мейснера [29]. Немиелинизирующие шванновские клетки, которые связаны с аксонами малого калибра,
называют ремаковскими (“Remak SC” [30]). При иннервации кожи, когда ремаковские нервные волокна доходят до эпидермиса,
их SCs достигают соотношения 1 шванновская клетка: 1 аксон. В дальнейшем эти волокна
теряют контакт со шванновскими клетками, и только аксоны проникают в эпидермис. В
отличие от миелинизированных аксонов, эти волокна характеризуются непрерывным ростом,
который находится под контролем фактора роста нервов, выделяемого немиелинизирующими
шванновскими клетками [30].
Немиелинизирующие шванновские клетки, которые связаны с аксонами в нервно-мышечных
контактах, называют терминальными или перисинаптическими шванновскими клетками [31, 32]. Осуществляют ли терминальные шванновские клетки трофическую функцию по отношению
к аксонам, как это делают ремаковские SCs, неизвестно. Предположительно они участвуют
в процессе синаптогенеза во время развития, а также в реиннервации нервных волокон
после травмы [27, 33, 34].
ПРОИСХОЖДЕНИЕ ШВАННОВСКИХ КЛЕТОК В ЭМБРИОГЕНЕЗЕ
Шванновские клетки происходят в эмбриогенезе из нервного гребня, закладки, которая
образуется во время замыкания нервной трубки и располагается в виде тяжей с двух сторон
от нее. Из мигрирующих мультипотентных клеток нервного гребня, наряду со шванновскими
клетками, образуются периферические нейроны, меланоциты, нейроэндокринные клетки и
др. [обзоры: 18, 25, 23, 35, 36].
После процесса миграции клетки нервного гребня проходят несколько стадий дифференцировки
в направлении шванновских клеток: предшественники (SCPs), незрелые шванновские клетки
(iSCs), немиелинизирующие шванновские клетки (pro-mSCs) и, наконец, дифференцированные,
среди которых одни миелинизируют аксоны, другие нет (mSCs и nmSCs) [23, 25]. Описание особенностей шванновских клеток и их фенотипа на разных этапах развития
обобщены в табл. 1.
Таблица 1.
Характеристика шванновских клеток на разных стадиях дифференцировки
| Стадия дифференцировки | Характеристика | Маркеры | Источ-ник |
|---|---|---|---|
| Предшественник шванновской клетки, Schwann cell precursor (SCP) |
Как и клетки нервного гребня, являются мигрирующими и способны к пролиферации. Их жизнеспособность зависит от сигналов, исходящих от растущих аксонов. SCP перемещаются вместе с развивающимися аксонами. Регулятор развития NRG1. Активация NRG1 ErbB2/3 имеет важное значение как для пролиферации SCP, так и для направленной миграции. |
Sox10, GAP43, Oct6, Sox2, MPZ | [23, 27, 37] |
| Незрелая шванновская клетка, immature Schwann cell (iSC) | Образуются из SCP и перестают мигрировать. Формируют базальную мембрану. Выживание iSC не зависит от аксональных факторов. В период их дифференцировки в нервах начинают формироваться сосуды и фибробласты, в экстрацеллюлярном матриксе появляется коллаген. Возникает периневральная оболочка. Механизмы, регулирующие этот этап дифференцировки, не вполне изучены, предполагается участие сигнального пути Notch. |
Sox10, S100, GAP43, P75NTR, NCAM, Sox2, Oct6, MPZ GFAP, O4 | [23, 27, 37] |
| Стадия промиелинизации, pro-myelin Schwann cell (pro-mSC) | Возникают на стадии, когда достигается соотношение 1 шванновская клетка: 1 аксон. Когда все аксоны большого калибра отделены. Pro-mSC образуют собственную базальную мембрану. Те незрелые SCs, которые охватывают оставшиеся аксоны небольшого калибра, дифференцируются в Remak SCs. |
Sox10, S100, Krox20, Oct6 | [27, 37] |
| Немиелинизирующая шванновская клетка, non-myelinating Schwann cell (nmSC) | Немиелинизирующие шванновские клетки окружают малые сенсорные и вегетативные аксоны ПНС, формируя классическое ремаковское волокно. Они сохраняют свои пролиферативные потенции. Терминальные шванновские клетки. Шванновские клетки телец Пачини и Мейснера. |
Sox10, S100, GAP43, P75NTR, NCAM, Oct6 Egr-1, GFAP и AN2/ NG2 | [27, 28, 37] |
| Миелинизирующая шванновская клетка, myelinating Schwann cell (mSC) | Шванновские клетки, образующие миелиновые оболочки нервных волокон большинства нервов. | Sox10, S100, Krox20, Oct6, MBP, MPZ P0/Pmp22/MAG/MBR | [18, 37] |
Предшественники шванновских клеток отличаются от клеток нервного гребня тем, что уже
находятся в тесной связи с аксонами. Есть данные, что они являются мультипотентными
клетками и способны дифференцироваться не только в шванновские клетки, но и в эндоневральные
фибробласты и ряд других типов клеток [23, 25]. Описывая эти клетки, авторы [23, 25] проводят параллель с клетками радиальной глии в развивающейся ЦНС, которые, являясь
нейральными стволовыми клетками, мультипотентны и дают начало нейронам, астроцитам,
эпендимным клеткам и олигодендроцитам. Молекулярный механизм дифференцировки клеток
нервного гребня в предшественники шванновских клеток мало изучен. Имеются лишь отдельные
данные о том, что в этом процессе участвует сигнальная система Notch [23]. Описана интересная особенность предшественников шванновских клеток при их культивировании:
в отсутствие аксонов они погибают.
Фенотипические характеристики шванновских клеток различаются в зависимости от стадии
их развития в онтогенезе. Маркеры шванновских клеток на разных этапах развития изучены
не только in vivo, но и in vitro. Liu et al. (2015) [37] с помощью иммунофлуоресценции, вестерн-блот анализа и количественной полимеразной
цепной реакции в реальном времени изучали специфические маркеры шванновских клеток
новорожденных мышей. Исследования проводились на культуре шванновских клеток. Были
выявлены десять маркеров, свойственных шванновским клеткам in vivo: S100, p75NTR, Sox10, Sox2, GAP43, NCAM, Krox20, Oct6, MBP и MPZ. На всех стадиях
развития были обнаружены только транскрипционные факторы Sox10 и Sox2. Через 8 сут
культивирования появлялись все маркеры, кроме GAP43 и Oct6. Было обнаружено, что широко
используемые маркеры S100 и P75NTR не были выражены на ранней стадии культивируемых
SCs.
Механизмы, контролирующие дифференцировку незрелых шванновских клеток в немиелинизирующие,
мало изучены. Есть данные, что в этом процессе участвует белок внеклеточного матрикса
– ламинин. Используя линию мутантных мышей, лишенных ламинина, Yu et al. (2009) [27, 28] показали, что необходимым стимулом для дифференцировки шванновских клеток является
именно этот белок.
Незрелые шванновские клетки (iSCs) выполняют важные гистогенетические функции. Во-первых,
они отделяют аксоны большого калибра, которые впоследствии станут миелинизированными,
от немиелинизированных волокон малого калибра. Во-вторых, они вырабатывают трофические
факторы, стимулирующие дифференцировку таких структурных элементов нерва, как периневральные
клетки, клетки кровеносных сосудов, фибробласты эндоневрия и эпиневрия [27].
ФУНКЦИИ ШВАННОВСКИХ КЛЕТОК
И ИХ ПРЕДШЕСТВЕННИКОВ
Миелинизация периферических нервных волокон
В процессе эволюции для увеличения скорости проводимости нервного импульса развивалось
два механизма: первый связан с увеличением диаметра аксона, второй – с возникновением
миелиновой оболочки [38]. Миелинизация позволила увеличить скорость проводимости аксонов малого калибра.
Миелиновые нервные волокна имеются у большинства позвоночных животных. Они появляются
в филогенезе у хрящевых рыб. Миелин акул и скатов устроен, как и у других позвоночных:
нервные волокна покрыты базальной пластинкой, окружены коллагеном, имеют много насечек
Штидта–Лантермана и перехваты Ранвье. Установлено, что как и у других позвоночных,
для миелиновых оболочек хрящевых рыб свойственны миелинпротеины P0, MPZ и основной
белок миелина (MBP) [39].
Что касается беспозвоночных животных, у представителей различных таксонов описан миелин
разного уровня развития. У одних – плотные миелиновые оболочки, у других – “голые”
нейриты, обернутые несколькими мембранами глиальных клеток [38, 40]. Так, у отдельных представителей ракообразных (крабы и омары), а также у моллюсков
и насекомых миелиновые оболочки не описаны. Изолирующую аксоны функцию здесь выполняют
немиелинизирующие глиальные клетки и соединительнотканные элементы. У тех представителей
ракообразных, которым свойственны миелиновые оболочки, например, у планктонных рачков
Copepoda, миелин имеет сходную структуру c таковой у позвоночных животных [41]. Их миелин характеризуется концентрически расположенными слоями мембраны вокруг
аксона. Количество слоев в оболочке изменяется для каждого аксона, варьируя от одного
до пятидесяти и более. Высокоорганизованные пластинки плотно обернуты вокруг заполненного
микротрубочками аксона. Оболочка состоит из электронноплотных слоев одинаковой толщины,
которые чередуются с менее плотными слоями большей толщины [41]. Такое сходство миелина позвоночных и беспозвоночных животных связано с идентичной
функцией миелинизирующих клеток.
Вопрос о том, какие именно условия среды привели к появлению миелина у беспозвоночных
животных, до сих пор не находит четкого объяснения [38]. Предположительно увеличение скорости проведения нервного импульса актуально для
тех животных, которым необходимо, скрываясь от хищников, или, наоборот, догоняя жертву,
развивать большую скорость движения [4, 38]. Подтверждением этой гипотезы является тот факт, что ареал обитания копепод, обладающих
миелинизированными волокнами, занимает большую территорию, чем ареал обитания копепод,
не имеющих миелинизированных волокон. Дискуссионным до сих пор является вопрос о том,
где первоначально в филогенезе появился миелин и миелинобразующие клетки: в центральной
или периферической нервной системе [42]. Некоторые исследователи считают, что SCs ПНС и олигодендроглия ЦНС имеют в эволюции
общего предшественника [2]. Однако между этими типами клеток множество различий. Например, различное эмбриональное
происхождение: олигодендроциты в эмбриогенезе происходят из нейроэктодермы, а шванновские
клетки – из нервного гребня. Между процессами миелинизации, проходящими по центральному
и периферическому типу, также много отличий. Их регулируют различные транскрипционные
каскады: Sox10, Olig1/2, MYRF для олигодендроглии и Sox10, Pou3F1, Egr2 для шванновских
клеток. Кроме того, как известно, у миелинизирующих клеток ЦНС и ПНС разные механизмы
“упаковки” миелина. Предполагается, что появление миелиновых структур в центральной
и периферической нервной системе происходило параллельно. Высказывается также мнение,
что миелин появлялся в эволюции несколько раз [4]. Имеются палеонтологические данные о том, что первыми позвоночными животными, которые
имели миелиновые нервные волокна, были панцирные рыбы [2, 3, 38].
В настоящее время активно изучаются молекулярные механизмы регуляции процесса миелинизации
периферических нервных проводников с применением различных моделей исследования. Среди
них культивирование нейронов и шванновских клеток in vitro, модели с применением трансгенных животных, модели повреждения нервов и др. [43].
Считается, что толщина миелиновой оболочки нервного волокна зависит от его диаметра
[8, 22]. В процессе формирования миелиновой оболочки необходимой толщины ключевую роль играет
передача сигналов NRG1 через рецепторы ErbB [43–45]. NRG1-сигнализация необходима шванновским клеткам для экспрессии структурных компонентов
цитоплазматической мембраны и осуществления нужного количества обвертываний вокруг
аксона. На моделях с применением трансгенных животных показано, что в отсутствие NRG1-III
миелинизации не происходит [18]. Доказательством значения NRG1 для осуществления миелинизации служит тот факт, что
немиелинизирующиеся в норме отростки симпатических нейронов становятся миелинизированными
в условиях in vitro, если такие нейроны обладают сверхэкспрессией NRG1 [24].
Трофическая функция шванновских клеток
Осуществление трофической функции миелинизирующих клеток (олигодендроцитов или SCs)
по отношению к нервной клетке и ее аксону ранее отмечалось многими авторами [1, 8, 22, 46], однако в наши дни при использовании современных методов исследования получены дополнительные
данные о влиянии глиальных клеток как ЦНС, так и ПНС, на метаболизм аксонов. Так,
показано, что олигодендроциты синтезируют лактат для аксонов и тем самым предотвращают
их дегенерацию [47]. Установлено, что нарушение функции митохондрий в SCs вызывает прогрессирующую дегенерацию
миелинизированных аксонов [48]. Важная роль в метаболизме аксонов отводится и немиелинизирующим шванновским клеткам
[27, 48].
Исследования взаимоотношений шванновских клеток и аксонов показали, что между ними
может осуществляться обмен органоидами. Имеются данные о том, что рибосомы могут переноситься
от SC к аксону при повреждении и в процессе развития [1]. Эксперименты, доказывающие перенос полирибосом из SCs в аксоны, были выполнены
с использованием зеленого флуоресцентного белка на моделях in vitro и in vivo. Рибосомы SCs были помечены зеленым флуоресцентным белком. После проникновения в
аксон меченые рибосомы можно было наблюдать в течение нескольких недель, где они предположительно
участвовали в местном синтезе белка [49]. Высказывается также мнение, что в нервах в области перехватов Ранвье или насечек
Шмидта-Лантермана может осуществляться процесс переноса РНК от SCs в аксоны [50].
Для ПНС трофическая функция глиальных клеток имеет особое значение, поскольку периферические
нервы достигают значительной длины, а транспорт метаболитов из тела нейрона осуществляется
медленно (по сравнению с транспортом везикул), их скорость составляет 300 мкм в час
[4].
Обмен генетической информацией
с аксоном путем высвобождения экзосом
Экзосомы и микровезикулы представляют собой внеклеточные нанопузырьки, высвобождаемые
многими клетками [51]. С их помощью осуществляются межклеточные взаимодействия путем передачи генетической
информации, включая передачу как кодирующих, так и некодирующих РНК, в клетки-реципиенты.
Экзосомы достигают размеров 10–100 нм. Показано, что экзосомы способны переносить
фрагменты матричной РНК (мРНК) и микроРНК (miRNA) из клетки в клетку [15, 51]. Изучая взаимоотношения между шванновской клеткой и аксоном, многие авторы показали,
что экзосомы, высвобождаемые SCs, влияют на регенерацию поврежденных аксонов. В обзорах
[50] и [52] дана характеристика экзосом и описаны механизмы их образования и значение в аксон-глиальных
взаимоотношениях.
В модельных экспериментах, выполненных in vitro, показано, что экзосомы шванновских клеток усваиваются аксонами периферических нервов,
и это приводит к стимуляции роста нейритов [52]. Установлено, что этот эффект специфичен для экзосом, полученных именно из шванновских
клеток. Доказательством служит тот факт, что экзосомы, синтезируемые не SCs, а фибробластами,
не оказывают такого влияния. Стимулирующее влияние экзосом шванновских клеток на рост
аксонов подтверждено и в экспериментах in vivo. Показано, что ежедневные инъекции экзосом в дистальный сегмент поврежденного нерва
приводят к двукратному увеличению скорости роста аксонов.
Предполагается, что экзосомы могут участвовать в регуляции валлеровской дегенерации
[50]. Известно, что экзосомы способны модулировать фенотип клеток путем переноса мРНК,
miRNAs и факторов транскрипции белков в различных органах [51]. Это позволяет предполагать, что с их помощью осуществляется переключение фенотипа
SCs с миелинизирующего зрелого на немиелинизирующий при дедифференцировке.
Синтез биологически активных веществ
Шванновские клетки оказывают влияние на нервные клетки, вырабатывая нейротрофические
факторы, цитокины и белки экстрацеллюлярного матрикса. Установлено, что они оказывают
поддерживающее влияние на развивающиеся нейроны, пока их аксоны еще не достигли органов-мишеней
[44]. Так, у трансгенных мышей с дефицитом ErbB3 (т.е. в отсутствие предшественников
SCs) двигательные нейроны спинного мозга и чувствительные нейроны спинномозговых ганглиев
погибают по механизму апоптоза [44].
После травмирования периферического нервного проводника и повреждения аксона SCs оказывают
поддерживающее влияние на нейроны, вырабатывая ряд биологически активных веществ:
фактор роста нервов (NGF), фактор роста фибро-бластов (FGF), нейротрофический фактор
головного мозга (BDNF), инсулиноподобный фактор роста (IGF), глиальный нейротрофический
фактор (GDNF), нейротрофин 4/5, нейротрофин 3(NT-3), цилиарный нейротрофический фактор,
нейрональный белок клеточной адгезии (NCAM) и др. [15, 53–56]. Доказательства необходимости этих факторов для сохранения жизнеспособности нейронов
и их дифференцировки были получены на моделях с использованием трансгенных животных.
Нарушение синтеза этих биологически активных веществ шванновскими клетками может приводить
к неврологическим заболеваниям и нарушению регенерации нервов.
Известно, что после повреждения нерва наблюдается дедифференцировка шванновских клеток.
Дедифференцированные SCs обретают свойство синтезировать ряд белков внеклеточного
матрикса (ламинина, фибронектина, тенасцина и др.), которые участвуют в стимуляции
регенерации аксонов [15, 27, 55]. Они секретируют также хемокины и цитокины, необходимые для привлечения в нерв моноцитов/макрофагов.
Среди них MCP-1 (моноцитарный хемоаттрактантный белок-1), LIF (leukemia inhibitory
factor), PAP-III (панкреатит-ассоциированный белок III) и интерлейкины IL-1α и IL-1β
[57–59].
Шванновские клетки способны синтезировать вещества, участвующие в регуляции морфогенетических
процессов, происходящих в нерве. Во-первых, они вырабатывают белок, который контролирует
образование периневрия – dhh (“desert hedgehog”) во время развития нерва [60]. Периневрий – одна из оболочек нервного ствола, которая обеспечивает периневральный
барьер, предотвращающий доступ в эндоневрий инфекционных агентов. Во-вторых, предшественники
шванновских клеток, ассоциированные с определенными нервами в период эмбриогенеза,
способны секретировать фактор роста эндотелия сосудов (VEGF-A), способствуя тем самым
процессам васкуло- и ангиогенеза в развивающемся нерве [61].
Таким образом, благодаря способности вырабатывать ряд биологически активных веществ
SCs оказывают трофическую поддержку развивающимся нейронам, пока их аксоны еще не
достигли мишеней, поддерживают жизнеспособность нервных клеток и целостность нервного
ствола после травмы нерва, а также участвуют в морфогенезе.
Участие в фагоцитозе продуктов распада миелина при валлеровской дегенерации
Процесс валлеровской дегенерации развивается в дистальном сегменте поврежденного нервного
ствола и включает в себя разрушение аксона и миелиновой оболочки. Считается, что валлеровская
дегенерация возникает в результате механической травмы нервных стволов, однако она
описана и при токсических или метаболических поражениях нервов [62]. Дегенерация осевого цилиндра нервного волокна приводит к нарушению его связи с
миелиновой оболочкой и вызывает разрушение миелина. Для осуществления нормальной регенерации
нервных волокон необходимо очищение нерва от продуктов распада аксонов и миелина.
Вопрос о том, какие клетки участвуют в уборке продуктов распада миелина, долгое время
считался дискуссионным [62, 8
]
. Роль фагоцитов, участвующих в уборке продуктов распада миелина в поврежденном нерве,
отводилась не только макрофагам, но и SCs. Существовало мнение, что шванновские клетки
миелинизированных волокон при повреждении освобождаются от миелина, и часть из них
участвует в фагоцитозе продуктов его распада. Доказательством этого служили результаты
электронномикроскопических исследований, в которых показано, что в цитоплазме SCs
после повреждения нерва наблюдаются миелин-ламмелярные структуры. Имелись также данные,
исключающие роль SCs в этом процессе. Например, на модели переживания фрагментов нерва
в диффузионной камере было установлено, что в условиях изоляции нерва от экзогенных
моноцитов/макрофагов фагоцитоз миелина не осуществляется [63]. В дальнейшем многими исследователями была доказана важная роль моноцитов/макрофагов
в устранении продуктов распада аксонов и миелина в поврежденных нервных проводниках
[22, 64, 65].
С появлением иммуногистохимических методов идентификации шванновских клеток и макрофагов
удалось установить, что в удалении продуктов распада миелина участвуют и одни, и другие
клетки [62]. В настоящее время не вызывает сомнения тот факт, что в течение первых суток после
травмы нерва функцию уборки продуктов распада миелина в его дистальном конце выполняют
шванновские клетки, а для окончательного очищения эндоневрия от миелина необходимо
подключение макрофагов [22, 62, 66]. В современных работах механизм устранения продуктов распада миелина шванновскими
клетками пересматривается. Высказывается мнение, что этим механизмом не может быть
фагоцитоз, поскольку при фагоцитозе поглощаются вещества, находящиеся вне клетки-фагоцита.
Миелиновая оболочка же является внутренним компонентом SC, ее частью. В связи с этим
предполагается, что механизм поглощения продуктов распада миелина шванновскими клетками
– макроавтофагия [67, 68]. Макроавтофагия является системой деградации, при которой клетки разрушают свои
собственные органеллы и крупные макромолекулы. Gomez‑Sanchez et al. (2015) [67] показали, что после повреждения нервного ствола в нем наблюдается активация аутофагии
SCs, фомируются аутофагосомы, содержащие продукты распада миелина. Авторы, наблюдая
аутофагию миелиновой оболочки SC, предложили для этого процесса термин “миелинофагия”.
На экспериментальных моделях показано, что генетическое и фармакологическое ингибирование
аутофагии приводит к ингибированию разрушения белков и липидов миелина в поврежденном
нерве.
ПЛАСТИЧНОСТЬ ШВАННОВСКИХ КЛЕТОК
О проявлении фенотипической пластичности шванновских клеток принято говорить, когда
происходит нарушение взаимодействия SCs с аксоном вследствие повреждения. Это нарушение
приводит к дедифференцировке как миелинизирующих, так и немиелинизирующих шванновских
клеток в незрелые формы.
После травмирования нерва зрелые шванновские клетки получают сигнал от поврежденного
аксона через систему NRG/Erg2 и меняют свой фенотип [22]. Показано, что Erg2 активируется в течение нескольких минут после травмы нервного
волокна, а в течение последующих двух суток после потери контакта с аксоном шванновские
клетки дедифференцируются [22]. Дедифференцированные клетки способны к миграции и пролиферации. Вследствие своего
деления они формируют в дистальном сегменте травмированного нерва так называемые “бюнгнеровские
ленты” – пути, по которым осуществляется рост регенерирующих аксонов. Кроме того,
они начинают синтезировать противовоспалительные цитокины и хемокины, стимулирующие
инфильтрацию макрофагов [59, 69]. Когда вследствие регенерации контакт с аксоном восстанавливается, эти клетки вновь
становятся миелинирующими или немиелинирующими в зависимости от сигнала, исходящего
от регенерирующего аксона [18]. В регуляции процессов дедифференцировки, пролиферации SCs и последующей ремиелинизации
регенерирующих аксонов участвуют белки внеклеточного матрикса, нейтрофические факторы
и гормоны [55]. Дедифференцированные шванновские клетки (клетки, формирующие “бюнгнеровские ленты”
в дистальном конце травмированного нерва) приобретают свойства незрелых SCs [70]. В исследованиях 2017 г. показано, что у них также имеются специфические черты,
отличные от других клеток в линии SCs. Gomez-Sanchez с коллегами [71] показали, что повреждение седалищных нервов у мышей приводит к изменению как структуры,
так и функции SCs. Эти клетки принимают удлиненную форму и образуют отростки. Показано,
что образующие “бюнгнеровские ленты” клетки в 2–3 раза длиннее миелинизирующих и ремаковских
шванновских клеток и в 7‒10 раз больше незрелых форм [71]. Примечательно, что когда эти клетки трансформируются обратно в миелинирующие клетки,
их размер уменьшается. Еще одной особенностью дедифференцированных шванновских клеток
является их способность оказывать стимулирующее влияние на репаративные процессы в
разных тканях [72].
Недавно показано, что предшественники шванновских клеток и клетки, формирующие “бюнгнеровские
ленты”, проявляют свойство мультипотентности. Так, в исследовании, выполненном на
модели восстановления резцов у мышей, установлено, что SCs генерируют клетки, из которых
образуются клетки пульпы и одонтобласты [73]. Экспериментально подтверждена возможность дифференцировки предшественников шванновских
клеток в меланоциты, клетки парасимпатических ганглиев нервной системы, мезенхимальные
стволовые клетки пульпы зуба [74]. Было продемонстрировано, что предшественники шванновских клеток в условиях in vitro в присутствии FGF-2 и эпидермального фактора роста могут быть перепрограммированы
в мультипотентные клетки, которые способны генерировать клетки, подобные нейронам,
глиоцитам и гладкомышечным клеткам [75]. Uesaka et al. (2015) [76] показали, что вегетативные нейроны энтеральной нервной системы могут формироваться
не только из клеток нервного гребня, но и из клеток-предшественников шванновских клеток.
РЕГЕНЕРАЦИЯ НЕРВА, ПАТОЛОГИЧЕСКАЯ ПРОЛИФЕРАЦИЯ ШВАННОВСКИХ КЛЕТОК И ДЕМИЕЛИНИЗИРУЮЩИЕ
ПРОЦЕССЫ
Изучение патологии нервных проводников, связанные с участием шванновских клеток (травма
нерва, демиелинизирующие расстройства, опухоли и др.), может пролить свет на молекулярные
механизмы регуляции дифференцировки SCs и их взаимоотношения с окружающими тканями.
После механической травмы нерва, которая может наступать при переломах, ушибах, сдавливании
близлежащей опухолью и др., SCs дедифференцируются и участвуют в репаративных процессах
и ремиелинизации. Однако часто нарушение последовательности и стройности этих процессов
приводит к необратимым изменениям, которые необходимо учитывать при хирургическом
лечении [15, 20]. В результате травмы периферического нервного проводника нередко возникает неврома,
которая препятствует нормальной репаративной регенерации нерва. Показано, что невромы
различных размеров могут возникать на проксимальном конце поврежденного нервного ствола,
сбоку от него и внутри ствола [7, 8]. Неврома состоит из большого числа миелинизированных и немиелинизированных нервных
волокон, периневральных футляров, эндоневральных фибробластов, шванновских клеток,
коллагеновых пучков соединительной ткани, нередко в ней встречаются кровоизлияния
и воспалительные инфильтраты [8]. Показано, что в невроме длительное время продолжаются процессы роста аксонов, миелинизиции,
ангиогенеза, а также пролиферация шванновских клеток и фибро-бластов, увеличение числа
макрофагов и тучных клеток [22]. Надо отметить, что морфофункциональные особенности шванновских клеток невромы сходны
с нормальными SCs в регенерирующем нерве. Неврому в эксперименте можно использовать
для изучения взаимоотношений клеточных элементов, составляющих нерв.
К демиелинизирующим заболеваниям относятся рассеянный склероз, болезнь Шарко-Мари-Тута
и синдром Гийена-Барре [18]. Болезнь Шарко-Мари-Тута является наследственным заболеванием и связана с дефектами
ключевых миелинизирующих генов. Синдром Гийена-Барре по своим проявлениям является
аутоиммунным расстройством. Демиелинизацию в нервной системе вызывает также Mycobacterium leprae, которые заражают SCs при проказе. Рецептор шванновских клеток альфа-дистрогликан
является сайтом связывания для M. leprae. Как только бактерии проникают внутрь клетки, М. leprae включаются в MAPKcascade для демиелинизации нервов и способствуют пролиферации SCs.
В результате наблюдается увеличение количества инфицированных клеток [77].
Среди опухолей нервных проводников различают доброкачественные (шваннома, нейрофиброма,
периневрома, травматическая неврома) и злокачественные (злокачественная опухоль периферической
нервной оболочки (malignant peripheral nerve sheath tumor (MPNST)) [78]. Шванномы возникают из миелинизирующих шванновских клеток и практически полностью
состоят из этих клеток. Нейрофибромы содержат все клеточные элементы периферического
нерва, включая SCs, фибробласты, периневриальные клетки и аксоны [78]. Различают нейрофиброматоз 1 типа (синдром Реклингхаузена) (NF1) и нейрофиброматоз
2 типа (NF2). Это аутосомно-доминантные наследственные заболевания. Опухоли в NF1
являются нейрофибромами и состоят из смеси фибробластов, шванновских клеток, периневриальных
клеток и тучных клеток [18]. Нейрофибромы иногда перерождаются в злокачественные опухоли периферических нервных
оболочек (MPNSTs или нейрофибросаркомы) [18].
Исследование патологии нервных стволов, в частности формирующихся в них опухолей,
позволяет изучать молекулярные механизмы регуляции тканевого гомеостаза в нерве. Так,
было выявлено, что одна из изоформ нейрегулина участвует в развитии нейрофибромы [79]. В отличие от нейрегулина III-1a, который ответственен за формирование миелиновых
волокон и за толщину миелиновой оболочки, тип нейрегулина III-3 не влияет на толщину
миелиновой оболочки. Используя трансгенных животных, показано, что сверхэкспрессия
этого белка у мышей приводит к увеличению размеров ганглиев и нервов. При этом они
приобретают признаки нейрофиброматоза I типа, о чем свидетельствует увеличение коллагеновых
волокон и количества шванновских клеток. При этом отмечается резкое изменение ремаковских
пучков: ремаковские шванновские клетки перестают отделять аксоны малого калибра друг
от друга. Вместо этого аксоны плотно упакованы, и весь пучок обернут SCs как единое
целое. Гиперпролиферация SCs и нарушение разделения аксонов в цитоплазме ремаковских
шванновских клеток также являются ранними признаками NF1 [79]. Полученные данные, по мнению авторов, подтверждают тот факт, что устойчивая активация
немиелинизирующих шванновских клеток нейрегулином, вырабатываемым аксоном, может способствовать
возникновению опухоли в ремаковских комплексах. Это указывает на то, что ингибирование
сигнальной передачи аксонов может служить в качестве предполагаемой терапии для лечения
опухолей ПНС [79].
В трех заболеваниях, связанных с пролиферацией SCs: проказа, NF1 и NF2, участвует
MAP-киназный каскад. Предполагается, что этот сигнальный путь играет центральную роль
в распространении заболевания по организму, поэтому некоторые исследователи считают,
что именно в этом направлении следует осуществлять поиск терапии этих заболеваний
[18] (наряду с антимикробной терапией, если это касается проказы). Из плексиформной нейрофибромы,
реже из шванномы [80], может развиваться MPNST. Клеточные и молекулярные особенности шванном были описаны
в 1920 г. шведским невропатологом Nils Ragnar Eugene Antoni [78]. Он описал два типа шванном, отличающихся по своей гистологии. В обзоре Wippold
et al. (2007) [78] охарактеризованы оба типа опухолевой ткани, носящие название Antoni A и B. Ткань
типа А является многоклеточной и содержит ламинин, ткань типа B состоит из большого
числа кистозных образований, кровеносных сосудов, областей некроза. Для шванном типа
А характерно высокое содержание белков базальных мембран: ламинина и коллагена IV.
Как известно, высокомолекулярный гликопротеин ламинин вырабатывается шванновскими
клетками на всех стадиях развития. Именно он является маркером, с помощью которого
можно дифференцировать опухоли, полученные из SCs, и отличать их от гистиоцитомы и
фибросаркомы [78]. Второй, свойственный SCs маркер, белок S100. Он также применяется для диагностики
опухолей. В шванномах он содержится практически во всех клетках, в нейрофибромах –
в некоторых, в MPNST – в единичных [78].
Исследование морфофункциональных особенностей SCs при различных заболеваниях позволяет
выявлять их гистобластические потенции в условиях измененного микроокружения. Исследование
молекулярных механизмов дифференцировки и уникальной пластичности этих клеток в опухолях
и при других заболеваниях нерва, а также изучение изменения сигнальных путей, регулирующих
эти процессы, имеет как общебиологическую значимость, так и большое практическое значение
для разработки подходов к лечению этих заболеваний.
ШВАННОВСКИЕ КЛЕТКИ И СТИМУЛЯЦИЯ РЕГЕНЕРАЦИИ НЕРВОВ
В литературе, посвященной разработке способов улучшения регенерации органов нервной
системы, шванновским клеткам уделяется значительное внимание. Это касается экспериментальных
работ, целью которых является восстановление спинного мозга или периферического нерва
после травмы.
Анализируя различные клеточные технологии, используемые для восстановления поврежденного
спинного мозга, Челышев и Викторов [81] обращают внимание на то, что после травмы спинного мозга в области его повреждения
появляются SCs. То есть в травмированном спинном мозге в процессе ремиелинизации аксонов
принимают участие SCs, а не только олигодендроглия (миелин-образующие клетки ЦНС).
Шванновские клетки мигрируют в область травмы спинного мозга из периферических нервных
структур при нарушении целостности барьера в результате повреждения. Высказываются
также предположения об участии резидентных нейральных клеток-предшественников спинного
мозга, дифференцирующихся в SCs, или о возможности олигодендроцитов спинного мозга
экспрессировать маркеры SCs при патологии. Главной целью применения клеточной терапии
при травме спинного мозга является восстановление или поддержание структуры и функции
именно белого вещества [81]. Здесь, в месте, где сосредоточены нервные волокна, использование трансплантации
SCs кажется уместным, ведь эти клетки формируют миелин в нервных проводниках. В оригинальных
исследованиях, выполненных десять лет назад как in vivo так и in vitro, установлено, что SCs способны миелинизировать аксоны ЦНС и улучшать их регенерацию.
В 90-е годы прошлого века были проведены первые работы по трансплантации экзогенных
шванновских клеток в поврежденные нервные стволы с целью улучшения их регенерации
[82]. Первоначально в искусственный кондуит, соединяющий концы перерезанного седалищного
нерва крыс линии с иммунодефицитом, наполненный матригелем, пересаживали SCs человека.
Используя антитела против клеток приматов, было установлено, что половина пересаженных
SCs выживает в течение 4 нед. Количество миелинизированных аксонов в кондуите было
значительно выше после применения клеточной терапии по сравнению с контролем. Некоторые
из трансплантированных человеческих шванновских клеток оказались способными миелинизировать
регенерирующие аксоны крысы. В то же время улучшение регенерации нерва было показано
после введения SCs в коллагеновый кондуит аллогенного животного [83]. Гистологический анализ показал, что SCs сохраняли свою жизнеспособность длительное
время (120 сут) и после трансплантации мигрировали на значительное расстояние от места
имплантации.
Позднее кондуиты, соединяющие проксимальный и дистальный сегменты нервов, становились
более совершенными [84]. Hadlock et al. [85] вводили SCs в кондуит из биодеградируемого материала (из высокомолекулярного сополимера
молочной и гликолевой кислот) с внутренними поверхностями, которые способствовали
прилипанию донорских SCs. Присутствие в такой конструкции шванновских клеток оказывало
стимулирующее влияние на рост аксонов. В дальнейшем исследования в этом направлении
проводились с использованием в качестве доноров животных, экспрессирующих зеленый
флуоресцентный белок (GFP), что позволило наблюдать за судьбой пересаженных клеток
[86, 87].
Как отмечалось ранее, шванновские клетки являются источником большого числа ростовых
и нейротрофических факторов, белков экстрацеллюлярного матрикса, цитокинов. Учитывая
эту особенность и необходимость биологически активных веществ, вырабатываемых SCs,
для восстановления поврежденных нервных волокон, появились экспериментальные работы,
в которых для трансплантации применялись не обычные SCs, а генетически модифицированные
по NGF, BDNF, FGF-2, GDNF или NT-3 [88–91]. Генная модификация SCs осуществляется с помощью плазмидного или вирусного векторов
[92]. В настоящее время в эксперименте применяется инъекция в поврежденный спинной мозг
животного непосредственно плазмиды или аденовируса с генами определенных факторов
роста (FRF2, VEGF, NDNF) [93, 94]. По данным некоторых авторов такой способ оказывается более эффективным для сохранения
нервных проводников спинного мозга, чем трансплантация генетически модифицированных
клеток [93, 94].
После того, как было установлено, что наполнение различного рода кондуитов (синтетических
и биологических) шванновскими клетками оказывает стимулирующее влияние на рост аксонов,
начались исследования по применению в качестве клеточной терапии различных стволовых
клеток, в частности мезенхимных стволовых клеток (MSCs) [обзоры: 9–16]. В течение
многих лет обсуждался вопрос о возможности MSCs или других используемых для пересадки
в нерв стволовых клеток дифференцироваться в SCs и миелинизировать регенерирующие
нервные волокна. С помощью иммуногистохимических маркеров и электронной микроскопии
в ряде работ было показано, что это возможно. Проводятся эксперименты по предифференцировке
MSCs, полученных из разных источников, в шванновские клетки в условиях in vitro перед тем, как вводить их в кондуит или непосредственно в поврежденный нерв.
Применение генной и клеточной терапии для восстановления нервных проводников в настоящее
время продолжает активно разрабатываться. Здесь хочется подчеркнуть, что не менее
важным направлением исследований является изучение реакции эндогенных шванновских
клеток нервных стволов на повреждение и применение клеточной терапии. Исследования
в этом направлении малочисленны. Есть данные о том, что повышение пролиферативной
активности SCs после применения экспериментальной клеточной терапии коррелирует с
улучшением роста аксонов [95]. Какие именно факторы, вырабатываемые пересаженными нейрогенными предшественниками,
являются митогенами для SCs, неясно. Не изучено также, увеличивается ли пролиферация
эндоневральных фибробластов при таких воздействиях. Предполагается, что ангиогенные
факторы, вырабатываемые MSCs, могут улучшать васкуляризацию нервных стволов [96]. Васкуляризация нерва имеет не только важное трофическое значение для его регенерации,
но способствует поступлению цитокинов и гормонов из кровеносного русла, которые оказывают
влияние на эндогенные клетки и на репаративные процессы в поврежденном нерве. Высказывается
мнение, что такие факторы роста как FGF или NGF, могут стимулировать пролиферацию
шванновских клеток. Учитывая роль SCs для регенерации аксонов, стимуляция именно эндогенных
шванновских клеток (а также других клеточных элементов нерва), на наш взгляд, может
быть новой терапевтической стратегией восстановления поврежденных нервных проводников.
ЗАКЛЮЧЕНИЕ
В настоящем обзоре обобщены имеющиеся в современной литературе данные о происхождении
шванновских клеток периферических нервных проводников в фило- и онтогенезе, а также
об их уникальной фенотипической пластичности. Описаны функции SCs, проявляющиеся на
разных стадиях их развития и после повреждения. Отмечены особенности SCs, выявляющиеся
при патологии периферических нервных проводников. В настоящее время на улучшение микроокружения
регенерирующих аксонов направлены новые стратегии применения биоинженерных конструкций,
соединяющих проксимальный и дистальный сегменты поврежденного нерва. Для этого используется
сочетание добавочных компонентов внеклеточного матрикса, нейротрофических факторов
и экзогенных стволовых и генетически модифицированных клеток, которые могут способствовать
росту и регенерации аксонов. Для применения новых клеточных и генных технологий в
качестве терапии поврежденного нерва необходимо более полное понимание молекулярно-клеточных
процессов, происходящих в регенерирующем нервном стволе. Малоизученными остаются молекулярные
факторы и сигнальные пути, участвующие в регенерации нервных волокон.
Дальнейшие исследования помогут понять, в каком направлении следует совершенствовать
клеточные и генные технологии восстановления нервных проводников, а также разобраться
в этиологии и патогенезе неврологических расстройств, связанных с дисфункцией шванновских
клеток.
Список литературы
-
Salzer J.L. Schwann cell myelination. Cold Spring Harb Perspect Biol. 7 (8): a020529. 2015.https://doi.org/10.1101/cshperspect.a020529
-
Zalc B. The acquisition of myelin: a success story. Novartis Found Symp. 276: 15–21. 2006.
-
Zalc B., Goujet D., Colman D. The origin of the myelination program in vertebrates. Curr. Biol. 18 (12): R511–512.
2008.https://doi.org/10.1016/j.cub.2008.04.010 -
Salzer J.L., Zalc B. Myelination. Curr. Biol. 26 (20): R971–R975. 2016.https://doi.org/10.1016/j.cub.2016.07.074
-
Waller A. New method for the study of the nervous system. Lond. J. Med. 4 (43): 609–625. 1852.
-
Ramon y Cajal, S. Degeneration and regeneration of the nervous system. 1–2. L.: Oxf. H. Milford. 1928.
-
Дойников Б.С. Избранные труды по нейроморфологии и невропатологии. М. 1955. [Doynikov B.S. Izbrannye trudy po nejromorfologii i nevropatologii. [The chosen works on neuromorphology
and neuropathology]. M. 1955 (in Russ)]. -
Ноздрачев А.Д., Чумасов Е.И. Периферическая нервная система. СПб. 1999. [Nozdrachev A.D., Chumasov E.I. Perifericheskaya nervnaya sistema [Peripheral nervous system]. SPb. 1999 (in Russ)].
-
Walsh S., Midha R. Use of stem cells to augment nerve injury repair. Neurosurgery. 65: 80–86. 2009.
-
Петрова Е. С. Изучение гистогенетических и нейродегенеративных процессов в нервной системе с помощью
гетеротопической нейротрансплантации. Морфология. 136 (6): 8–19. 2009. [Petrova E. S. Studies of the histogenetic and neurodegenerative processes in the nervous system
using heterotopic neurotransplantation. Morphology. 136 (6): 8–19. 2009 (in Russ)]. -
Челышев Ю.А. Регенерация в нервной системе. Руководство по гистологии / Под ред. Р.К. Данилова.
С. 656–665. СПб.: СпецЛит. 2011. [Chelyshev Yu.A. Regeneraciya v nervnoj sisteme. Rukovodstvo po gistologii [Regeneration in nervous
system. The guide on histology] / Ed. R.K. Danilov. P. 656–665. SPb.: SpecLit. 2011
(in Russ)]. -
Петрова Е.С. Применение стволовых клеток для стимуляции регенерации поврежденного нерва. Цитология.
54: 525–540. 2012. [Petrova E.S. The use of stem cells to stimulate regeneration of damaged nerve. Cytology. 54: 525–540.
2012 (in Russ)]. -
Петрова Е.С. Восстановление поврежденного нерва с помощью клеточной терапии (фундаментальные аспекты).
Acta Naturae (русскоязычная версия). 7 (3 (26)): 42–53. 2015. [Petrova E.S. Injured nerve regeneration using cell-based therapies: current challenges. Acta Naturae
7 (3 (26)): 42–53. 2015 (in Russ)]. -
Fairbairn N.G., Meppelink A.M., Ng-Glazier J., Randolph M.A., Winograd J.M. Augmenting peripheral nerve regeneration using stem cells: A review of current opinion.
World J. Stem Cells. 7 (1): 11–26. 2015. -
Щаницын И.Н., Иванов А.Н., Бажанов С.П., Нинель В.Г., Пучиньян Д.М., Норкин И.А. Cтимуляция регенерации периферического нерва: современное состояние, проблемы и перспективы.
Успехи физиол. наук. 48 (3): 92–112. 2017. [Shchanitsyn I.N., Ivanov A.N., Bazhanov S.P., Ninel’ V.G., Puchin’jan D.M., Norkin
I.A. Stimulation of Peripheral Nerve Regeneration: Current Status, Problems and Perspectives.
Uspekhi Fiziologicheskikh Nauk [Progress in the Physiological Sciences] 48 (3): 92–112.
2017 (in Russ)]. -
Карагяур М.Н., Макаревич П.И., Шевченко Е.К., Стамбольский Д.В., Калинина Н.И., Парфенова Е.В. Современные подходы к регенерации периферических нервов: перспективы генной и клеточной
терапии. Гены и клетки. 12 (1): 6–14. 2017. [Karagyaur M.N., Makarevich P.I., Shevchenko E.K., Stambolsky D.V., Kalinina N.I.,
Parfyonova Ye.V. Modern approaches to peripheral nerve regeneration after injury: the prospects of
gene and cell therapy. Genes and Cells. 12 (1): 6–14. 2017 (in Russ)]. -
Шванн Т. Микроскопические исследования о соответствии в структуре и росте животных и растений.
М.-Л. 1939. [Schwann T. Microscopical researches into the accordance in the structure and growth of animals
and plants. London. 1847. (Russ Ed.: Schwann T. Mikroskopicheskie issledovaniya o sootvetstvii v strukture i roste zhivotnyh i rastenij.
M-L. 1939)]. -
Bhatheja K., Field J. Schwann cells: origins and role in axonal maintenance and regeneration. Int. J. Biochem.
Cell Biol. 38: 1995–1999. 2006. -
Pineda A. The “lemmocyte” in peripheral-nerve tumors. J. Neurosurg. 22: 594–601. 1965.
-
Одинак М.М., Живолупов С.А., Рашидов Н.А., Самарцев И.Н. Особенности развития дегенерационно-реиннервационного процесса при травматических
невропатиях и плексопатиях. Вестник Российской Военно-медицинской академии. 4 (20):
130–140. 2007. [Odinak M.M., Zhivolupov S.A., Rashidov N.A., Samartsev I.N. Peculiarities of development of denervation-reinervation process in traumatic neuropathies
and plexopathies. Bulletin of the Russian Military Medical Academy. 4 (20): 130–140.
2007. (in Russ)]. -
Terminologia histologica. Международные термины по цитологии и гистологии человека
с официальным списком русских эквивалентов / Под ред. В. В. Банина, В. Л. Быкова.
М. Из-во: ГЭОТАР-Медиа. 2009. [Terminologia histologica. Mezhdunarodnye terminy po
citologii i gistologii cheloveka s oficial’nym spiskom russkih ekvivalentov [Terminologia
histologica. The international terms of the human cytology and histology with the
official list of the Russian equivalents] / Eds. V.V. Banin, V. L. Bykov. M.: GEOTAR-Media.
2009 (in Russ)]. -
Zochodne D. W. Neurobiology of peripheral nerve regeneration. Cambridge, New York, Melbourne, Madrid,
Cape Town, Singapore, Sao Paulo, 2008. -
Jessen K.R., Mirsky R., Lloyd A.C. Schwann cells: development and role in nerve repair. Cold Spring Harb. Perspect.
Biol. 7 (7): a020487. 2015.https://doi.org/10.1101/cshperspect.a020487 -
Taveggia C., Zanazzi G., Petrylak A., Yano H., Rosenbluth J., Einheber S., Xu X.,
Esper R.M., Loeb J.A., Shrager P., Chao M.V., Falls D.L., Role L., Salzer J.L. Neuregulin-1 type III determines the ensheathment fate of axons. Neuron. 47: 681–694.
2005. -
Jessen K.R., Mirsky R., Lloyd A.C. Schwann cells: development and role in nerve repair. Cold Spring Harb. Perspect.
Biol. 2015. 7 (7): a020487. Michailov G.V., Sereda M.W., Brinkmann B.G., Fischer
T.M., Haug B., Birchmeier C., Role L., Lai C., Schwab M.H., Nave K.A. Axonal neuregulin-1
regulates myelin sheath thickness. Science. 304: 700–703. 2004.https://doi.org/10.1101/cshperspect.a020487 -
Zalc B. The acquisition of myelin: An evolutionary perspective. Brain Research. 1641 (Pt
A): 4–10. 2016. -
Monk K.R., Feltri M.L., Taveggia C. New insights on Schwann cell development. Glia. 63: 1376–1393. 2015.
-
Yu W.M., Yu H., Chen Z.L., Strickland S. Disruption of laminin in the peripheral nervous system impedes nonmyelinating Schwann
cell development and impairs nociceptive sensory function. Glia. 57: 850–859. 2009. -
Griffin J.W., Thompson W.J. Biology and pathology of nonmyelinating Schwann cells. Glia. 56: 1518–1531. 2008.
-
Diamond J., Holmes M., Coughlin M. Endogenous NGF and nerve impulses regulate the collateral sprouting of sensory axons
in the skin of the adult rat. J. Neurosci. 12: 1454–1466. 1992. -
Young P., Nie J., Wang X., McGlade C.J., Rich M.M., Feng G. LNX1 is a perisynaptic Schwann cell specific E3 ubiquitin ligase that interacts with
ErbB2. Mol. Cell. Neurosci. 30: 238–248. 2005. -
Zuo Y., Lubischer J.L., Kang H., Tian L., Mikesh M., Marks A., Scofield V.L., Maika
S., Newman C., Krieg P., Thompson W.J. Fluorescent proteins expressed in mouse transgenic lines mark subsets of glia, neurons,
macrophages, and dendritic cells for vital examination. J. Neurosci. 24: 10999–11009.
2004. -
Smith I.W., Mikesh M., Lee Y., Thompson W.J. Terminal Schwann cells participate in the competition underlying neuromuscular synapse
elimination. J. Neurosci. 33: 17724–17736. 2013. -
Kang H., Tian L., Mikesh M., Lichtman J.W., Thompson W.J. Terminal Schwann cells participate in neuromuscular synapse remodeling during reinnervation
following nerve injury. J. Neurosci. 34: 6323–6333. 2014. -
Le Douarin N.M. Cell line segregation during peripheral nervous system ontogeny. Science. 231: 1515–1522.
1986. -
Kidd G.J., Ohno N., Trapp B.D. Biology of Schwann cells. Handb. Clin. Neurol. 115: 55–79. 2013.
-
Liu Z., Jin Y.Q., Chen L., Wang Y., Yang X., Cheng J., Wu W., Qi Z., Shen Z. Specific marker expression and cell state of Schwann cells during culture in vitro.
PLoS One. 10 (4): e0123278. 2015.https://doi.org/10.1371/journal.pone.0123278 -
Hartline D.K., Colman D.R. Rapid conduction and the evolution of giant axons and myelinated fibers. Curr Biol.
17 (1): R29-35. 2007.https://doi.org/10.1016/j.cub.2006.11.042 -
De Bellard M.E. Myelin in cartilaginous fish. Brain Res. 1641 (Pt A): 34–42. 2016.
-
Hartline D.K. The evolutionary origins of glia. Glia. 59: 1215–1236. 2011.
-
Davis A.D., Weatherby T.M., Hartline D.K., Lenz P.H. Myelin-like sheaths in copepod axons. Nature. 398: 571. 1999.
-
Kastriti M.E., Adameyko I. Specification, plasticity and evolutionary origin of peripheral glial cells. Curr.
Opin. Neurobiol. 47: 196–202. 2017. -
Birchmeier C. ErbB receptors and the development of the nervous system. Exp. Cell Res. 315: 611–618.
2009. -
Riethmacher D., Sonnenberg-Riethmacher E., Brinkmann V., Yamaai T., Lewin G.R., Birchmeier
C. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature.
389: 725–730. 1997. -
Nave K.-A., Trapp B.D. Axon-glial signaling and the glial support of axon function. Annu. ReNeurosci. 31:
535–561. 2008. -
Varon S.S., Bunge R.P. Trophic mechanisms in the peripheral nervous system. Annu. ReNeurosci. 1: 327–361.
1978. -
Morrison B.M., Lee Y., Rothstein J.D. Oligodendroglia: metabolic supporters of axons. Trends Cell Biol. 23: 644–651. 2013.
-
Viader A., Sasaki Y., Kim S., Strickland A., Workman C.S., Yang K., Gross R.W., Milbrandt
J. Aberrant Schwann cell lipid metabolism linked to mitochondrial deficits leads to
axon degeneration and neuropathy. Neuron. 77: 886–898. 2013. -
Court F.A., Midha R., Cisterna B.A., Grochmal J., Shakhbazau A., Hendriks W.T., Van
Minnen J. Morphological evidence for a transport of ribosomes from Schwann cells to regenerating
axons. Glia. 59: 1529–1539. 2011. -
Ching R.C., Kingham P.J. The role of exosomes in peripheral nerve regeneration. Neural Regen. Res. 10: 743–747.
2015. -
Lee Y., El Andaloussi S., Wood M.J. Exosomes and microvesicles: extracellular vesicles for genetic information transfer
and gene therapy. Human Molecular Genetics. 21 (R1): R125–134. 2012.https://doi.org/10.1093/hmg/dds317 -
Lopez-Leal R., Court F.A. Schwann cell exosomes mediate neuron-glia communication and enhance axonal regeneration.
Cell Mol. Neurobiol. 36: 429–436. 2016. -
Heumann R. Regulation of the synthesis of nerve growth factor. J. Exp. Biol. 132: 133–150. 1987.
-
Lewin G R., Barde Y.A. Physiology of the neurotrophins. Annu. ReNeurosci. 19: 289–317. 1996.
-
Chen Z.L., Yu W.M., Strickland S. Peripheral regeneration. Annu. ReNeurosci. 30: 209–233. 2007.
-
Jiang X., Liu L., Zhang B., Lu Z ., Qiao L., Feng X., Yu W. Effects of Angelica extract on schwann cell proliferation and expressions of related
proteins. Evid Based Complement Alternat Med. 2017: 6358392. 2017.https://doi.org/10.1155/2017/6358392 -
Shamash S., Reichert F., Rotshenker S. The cytokine network of Wallerian degeneration: tumor necrosis factoralpha, interleukin-1alpha,
and interleukin-1beta. J. Neurosci. 22: 3052–3060. 2002. -
Tofaris G.K., Patterson P.H., Jessen K.R., Mirsky R. Denervated Schwann cells attract macrophages by secretion of leukemia inhibitory
factor (LIF) and monocyte chemoattractant protein-1 in a process regulated by interleukin-6
and LIF. J. Neurosci. 22: 6696–6703. 2002. -
Chen P., Piao X., Bonaldo P. Role of macrophages in Wallerian degeneration and axonal regeneration after peripheral
nerve injury. Acta Neuropathol. 130: 605–618. 2015. -
Jung J., Frump D., Su J., Wang W., Mozaffar T., Gupta R. Desert hedgehog is a mediator of demyelination in compression neuropathies. Exp.
Neurol. 271: 84–94. 2015. -
Li W., Kohara H., Uchida Y., James J.M., Soneji K., Cronshaw D.G., Zou Y.R., Nagasawa
T., Mukouyama Y.S. Peripheral nerve-derived CXCL12 and VEGF-A regulate the patterning of arterial vessel
branching in developing limb skin. DeCell. 24: 359–371. 2013. -
Hirata K., Kawabuchi M. Myelin phagocytosis by macrophages and nonmacrophages during Walleriandegeneration.
Microsc. Res. Tech. 57: 541–547. 2002. -
Beuche W., Friede R.L. The role of non-resident cells in Wallerian degeneration. J. Neurocytol. 13: 767–796.
1984. -
Чумасов Е.И., Светикова К.М. Строение и природа макрофагов, участвующих в процессе уоллеровской дегенерации нервных
волокон. Архив анатомии, гистологии и эмбриологии. Т. 100. № 5. С. 13–21. 1991. [Chumasov E.I., Svetikova K.M. The structure and nature of the macrophages participating in Wallerian degeneration
of nerve fibers. Arkh Anat Gistol Embriol. 100 (5): 13–21. 1991 (in Russ)]. -
Brosius Lutz A., Barres B.A. Contrasting the glial response to axon injury in the central and peripheral nervous
systems. DeCell. 28: 7–17. 2014. -
Perry V.H., Tsao J.W., Fearn S., Brown M.C. Radiation-induced reductions in macrophage recruitment have only slight effects on
myelin degeneration in sectioned peripheral nerves of mice. Eur. J. Neurosci. 7: 271–280.
1995. -
Gomez-Sanchez J.A., Carty L., Iruarrizaga-Lejarreta M., Palomo-Irigoyen M., Varela-Rey
M., Griffith M., Hantke J., Macias-Camara N., Azkargorta M., Aurrekoetxea I., De Juan
V.G., Jefferies H.B., Aspichueta P., Elortza F., Aransay A.M., Martнnez-Chantar M.L.,
Baas F., Mato J.M., Mirsky R., Woodhoo A., Jessen K.R. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves.
J. Cell Biol. 210: 153–168. 2015. -
Brosius Lutz A., Chung W.S., Sloan S.A., Carson G.A., Zhou L., Lovelett E., Posada
S., Zuchero J.B., Barres B.A. Schwann cells use TAM receptor-mediated phagocytosis in addition to autophagy to
clear myelin in a mouse model of nerve injury. Proc. Natl. Acad. Sci. U S A. 114 (38):
E8072-E8080. 2017.https://doi.org/10.1073/pnas.1710566114 -
Cámara-Lemarroy C.R., Guzmán-de la Garza F.J., Fernández-Garza N.E. Molecular inflammatory mediators in peripheral nerve degeneration and regeneration.
Neuroimmunomodulation. 17: 314–324. 2010. -
Arthur-Farraj P.J., Latouche M., Wilton D.K., Quintes S., Chabrol E., Banerjee A.,
Woodhoo A., Jenkins B., Rahman M., Turmaine M., Wicher G.K., Mitter R., Green-smith
L., Behrens A., Raivich G., Mirsky R., Jessen K.R. C-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential
for regeneration. Neuron. 75: 633–647. 2012. -
Gomez-Sanchez J.A., Pilch K.S., van der Lans M., Fazal S.V., Benito C., Wagstaff L.J.,
Mirsky R., Jessen K.R. After Nerve Injury, Lineage Tracing Shows That Myelin and Remak Schwann Cells Elongate
Extensively and Branch to Form Repair Schwann Cells, Which Shorten Radically on Remyelination.
J. Neurosci. 37: 9086–9099. 2017. -
Carr M.J., Johnston A.P. Schwann cells as drivers of tissue repair and regeneration. Curr. Opin. Neurobiol.
47: 52–57. 2017. -
Kaukua N., Shahidi M.K., Konstantinidou C., Dyachuk V., Kaucka M., Furlan A., An Z.,
Wang L., Hultman I., Ahrlund-Richter L., Blom H., Brismar H., Lopes N.A., Pachnis
V., Suter U., Clevers H., Thesleff I., Sharpe P., Ernfors P., Fried K., Adameyko I. Glial origin of mesenchymal stem cells in a tooth model system. Nature. 513: 551–554.
2014. -
Adameyko I., Lallemend F., Aquino J.B., Pereira J.A., Topilko P., Mü ller T., Fritz N., Beljajeva A., Mochii M., Liste I., Usoskin D., Suter U., Birchmeier
C., Ernfors P. Schwann cell precursors from nerve innervation are a cellular origin of melanocytes
in skin. Cell. 139: 366–379. 2009. -
Widera D., Heimann P., Zander C., Imielski Y., Heidbre-der M., Heilemann M., Kaltschmidt
C., Kaltschmidt B. Schwann cells can be reprogrammed to multipotency by culture. Stem Cells Dev. 20:
2053–2064. 2011. -
Uesaka T., Nagashimada M., Enomoto H. Neuronal Differentiation in Schwann cell lineage underlies postnatal neurogenesis
in the enteric nervous system. J. Neurosci. 35: 9879–9888. 2015. -
Tapinos N., Rambukkana A. Insights into regulation of human Schwann cell proliferation by Erk1/2 via a MEK-independent
and p56Lck-dependent pathway from leprosy bacilli. Proc. Natl. Acad. Sci. U S A. 102:
9188–9193. 2005. -
Wippold F.J. 2nd, Lubner M., Perrin R.J., Lдmmle M., Perry A. Neuropathology for the neuroradiologist: Antoni A and Antoni B tissue patterns. AJNR
Am. J. Neuroradiol. 28 (9): 1633–1638. 2007. -
Gomez-Sanchez J.A., Lopez de Armentia M., Lujan R., Kessaris N., Richardson W.D.,
Cabedo H. Sustained axon-glial signaling induces Schwann cell hyperproliferation, Remak bundle
myelination, and tumorigenesis. J. Neurosci. 29: 11304–11315. 2009. -
Ненашев Е.А., Черекаев В.А., Кадашева А.Б., Козлов А.В., Ротин Д.Л., Степанян М.А.Трансформация нейрофибромы первой ветви тройничного нерва в злокачественную опухоль
оболочек периферических нервов (MPNST). Вопросы нейрохирургии им. Н.Н. Бурденко. 76
(5): 58–62. 2012. [Nenashev E.A., Cherekaev V.A., Kadasheva A.B., Kozlov A.V., Rotin D.L., Stepanian
M.A. Transformation of trigeminal nerve tumor into malignant peripheral nerve sheath tumor
(MPNST). Problems of Neurosurgery 76 (5): 58–62. 2012. (in Russ)]. -
Челышев Ю.А., Викторов И.В. Клеточные технологии ремиелинизации при травме спинного мозга. Неврологический вестник.
41 (1): 49–55. 2009. [Chelishev J.A., Victorov I.V. Cellular technologies of remyelinization at spinal cord trauma. Nevrologicheskiy
vestnik [Neurologic bulletin] 41 (1): 49–55. 2009 (in Russ)]. -
Levi A.D., Gunard V., Aebischer P., Bunge R.P. The functional characteristics of Schwann cells cultured from human peripheral nerve
after transplantation into a gap within the rat sciatic nerve. J. Neurosci. 14 (3
Pt 1): 1309–1319. 1994. -
Kim D.H., Connolly S.E., Kline D.G., Voorhies R.M., Smith A., Powell M., Yoes T.,
Daniloff J.K. Labeled Schwann cell transplants versus sural nerve grafts in nerve repair. J. Neurosurg.
80: 254–260. 1994. -
Rajangam T., An S.S. Fibrinogen and fibrin based micro and nano scaffolds incorporated withdrugs, proteins,
cells and genes for therapeutic biomedical applications. Int. J. Nanomedicine. 8:
3641–3662. 2013. -
Hadlock T., Sundback C., Hunter D., Cheney M., Vacanti J.P. A polymer foam conduit seeded with Schwann cells promotes guided peripheral nerve
regeneration. Tissue Eng. 6: 119–127. 2000. -
Radtke C., Akiyama Y., Lankford K.L., Vogt P.M., Krause D.S., Kocsis J.D.Integration of engrafted Schwann cells into injured peripheral nerve: axonal association
and nodal formation on regenerated axons. Neurosci Lett. 387: 85–89. 2005. -
Schmitte R., Tipold A., Stein V.M., Schenk H., Flieshardt C., Grothe C., Haastert
K. Genetically modified canine Schwann cells–In vitro and in vivo evaluation of their
suitability for peripheral nerve tissue engineering. J. Neurosci. Methods. 186: 202–208.
2010. -
Timmer M., Robben S., Mü ller-Ostermeyer F., Nikkhah G., Grothe C. Axonal regeneration across long gaps in silicone chambers filled with Schwann cells
overexpressing high molecular weight FGF-2. Cell Transplant. 12: 265–277. 2003. -
Haastert K., Lipokatic E., Fischer M., Timmer M., Grothe C. Differentially promoted peripheral nerve regeneration by grafted Schwann cells over-expressing
different FGF-2 isoforms. Neurobiol. Dis. 21: 138–53. 2006. -
Li Q., Ping P., Jiang H., Liu K. Nerve conduit filled with GDNF gene-modified Schwann cells enhances regeneration
of the peripheral nerve. Microsurgery. 26: 116–121. 2006. -
Pettingill L.N., Minter R.L., Shepherd R.K. Schwann cells genetically modified to express neurotrophins promote spiral ganglion
neuron survival in vitro. Neuroscience. 152: 821–818. 2008. -
Madduri S., Gander B. Schwann cell delivery of neurotrophic factors for peripheral nerve regeneration.
J. Peripher. Nerv. Syst. 15: 93–103. 2010. -
Шаймарданова Г.Ф., Башанкаев С.Д., Измайлов А.А., Фадеев Ф.О., Соколов М.Е., Исламов
Р.Р. Стимуляция регенерации спинного мозга крысы аденовирусами, содержащими гены, кодирующие
GDNF, NCAM1, VEGF165. Морфология. 151 (3): 115. 2017. [Shaymardanova G.F., Bashankayev S.D., Izmaylov A.A., Fadeyev F.O., Sokolov M. Ye.,
Islamov R.R. Stimulation of regeneration of rat spinal cord by adenoviruses carrying genes encoding
GDNF, NCAM1 AND VEGF165. Morphology. 151 (3): 115. 2017 (in Russ)]. -
Шаймарданова Г.Ф., Мухамедшина Я.О., Челышев Ю.А. Оценка эффективности путей локальной доставки терапевтических генов при травме спинного
мозга крысы: корреляции параметров структуры и функции. Современные технологии в медицине.
5 (3): 16–22. 2013. [Shaymardanova G.F., Mukhamedshina Y.О., Chelyshev Y.А. The Assessment of Efficiency of Local Delivery Pathways of Therapeutic Genes in Murine
Spinal Cord Injury: Correlation of Structure and Function Parameters. Sovremennye
tehnologii v medicine [Modern Technologies in Medicine]. 5 (3): 16–22. 2013. (in Russ)]. -
Петрова Е.С. Изучение регенерации передавленного седалищного нерва крысы после применения экспериментальной
клеточной терапии. Международный научно-исследовательский журнал. 4 (70): 42–45. 2018.
[Petrova E.S. Study on regeneration of crushed sciatic nerve of rat after use of experimental cell
therapy. Mezhdunarodnyj nauchno-issledovatel’skij zhurnal [International scientific
research journal]. 4 (70): 42–45. 2018. (in Russ)]. -
Петрова Е.С., Исаева Е.Н., Колос Е.А., Коржевский Д.Э. Васкуляризация поврежденного нерва крысы после применения экспериментальной клеточной
терапии. Клеточные технологии в биологии и медицине. 1: 53–57. 2018. [Petrova E.S., Isaeva E.N., Kolos E.A., Korzhevskii D.E. Vascularization of the Damaged Nerve under the Effect of Experimental Cell Therapy.
Cell Technologies in Biology and Medicine. 1: 53–57. 2018. (in Russ)].
Дополнительные материалы отсутствуют.
Schwann Cell Morphology☆
A.A. Lavdas, R. Matsas, in Reference Module in Biomedical Sciences, 2014
Abstract
Schwann cells (SCs) are the major glial cell type in the peripheral nervous system. They play essential roles in the development, maintenance, function, and regeneration of peripheral nerves. In the mature nervous system, SCs can be categorized into two major classes: myelinating and nonmyelinating cells. Myelinating SCs provide the myelin ensheathment of all large-diameter peripheral axons. Each myelinating SC associates with a single axon. Nonmyelin-forming SCs associate with smaller axons in such a manner that one SC enfolds several axons to form a Remak bundle. This article discusses the morphological characteristics of SCs and how they affect the architecture of peripheral nerves.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128012383047589
Schwann Cells and Axon Relationship
R.D. Fields, in Encyclopedia of Neuroscience, 2009
Embryological Origin of Schwann Cells
Schwann cells derive embryologically from the neural crest, which comprises multipotent cells migrating away from the dorsal neural tube. Neural crest cells differentiate into Schwann cell precursors, which migrate and proliferate along tracts of axons that have already extended into the periphery. The Schwann cell precursors then embark on a process of cellular differentiation into an immature Schwann cell to eventually become either a myelinating or a nonmyelinating Schwann cell. Only large-diameter axons, which conduct impulses at the highest speed, become myelinated. The slender, slow conducting fibers become bundled together and engulfed by massive, globular nonmyelinating Schwann cells.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780080450469006987
Spinal Cord Injury
Mary Bartlett Bunge, Patrick Mcghee Wood, in Handbook of Clinical Neurology, 2012
Polysialylated Neural Cell Adhesion Molecule-Engineered Schwann Cells Promote More Repair Than Control Schwann Cells
SC myelination in a compression injury model was improved using a strategy to promote SC migration by changing implanted SC surface adhesivity (Papastefanaki et al., 2007). The polysialylated (PSA) form of the neural cell adhesion molecule, NCAM, is present on SCs acutely after spinal cord demyelination but disappears as myelin repair is accomplished (Nait Oumesmar et al., 1995). Thinking that PSA would be beneficial, early postnatal GFP-SCs were engineered to express on their surface the PSA form of NCAM by introducing a retroviral vector encoding sialyl-transferase X (STX) which catalyzes transfer of PSA to NCAM (Papastefanaki et al., 2007). In vitro, 11X more STX-GFP-SCs entered astrocyte domains than did control SCs. In vivo, STX-GFP-SCs injected just rostral to a mouse thoracic compression injury promoted faster and significantly greater functional recovery (Basso mouse scale (BMS); Basso et al., 2006) compared to control engineered SCs or no cells. The earlier improvement in locomotor recovery in the STX-GFP-SC group was perhaps related to the earlier enhanced myelination by the implanted STX-GFP-SCs, increased myelination by host SCs, and enhanced recruitment/myelination by oligodendrocyte precursors. In addition, more serotonergic axons were observed to exit the caudal lesion site in the STX-GFP-SC group than in the control-GFP-SC group. Overall, STX-GFP-SC grafting appeared to lead to a more permissive CNS milieu for axonal growth and myelination, resulting in locomotor improvement (Papastefanaki et al., 2007).
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780444521378000322
Signaling Pathways that Regulate Glial Development and Early Migration – Schwann Cells
K.R. Jessen, R. Mirsky, in Patterning and Cell Type Specification in the Developing CNS and PNS, 2013
40.4 Major Differences Among Migrating Neural Crest Cells, SCPs, and iSchs
SCPs within embryonic nerves differ from migrating neural crest cells because of several reasons: first, because they express glial differentiation markers and other factors not found on migrating crest cells (Figure 40.2; Buchstaller et al., 2004; D’Antonio et al., 2006b; Li et al., 2007; Mirsky et al., 2008). Second, as mentioned earlier, SCPs exhibit the diagnostic glial phenotype of intimate association with nerve cells as they are found among axons within nerves, while crest cells migrate through mesenchymal connective tissue. Third, SCPs and crest cells respond differently to survival signals and mitogens (Woodhoo et al., 2004). Lastly, compared to crest cells, SCPs are insensitive to the neurogenic action of BMP2 and strongly biased toward the generation of iSchs (Kubu et al., 2002; Woodhoo and Sommer, 2008). For further discussion of the neural crest, see Chapter 20.

Figure 40.2. The phenotype of the main stages of the embryonic Schwann cell lineage. Below the lineage drawing are some of the molecular markers that characterize the lineage. Four groups are shown: (i) markers that show no significant change between the three stages, (ii) markers that are upregulated at the crest/SCP transition, (iii) markers that are upregulated at the SCP/iSch transition, and (iv) markers that are downregulated at the SCP/iSch transition.
For references, see text and Jessen KR and Mirsky R (2005) The origin and development of glial cells in peripheral nerves. Nature Reviews Neuroscience 6: 671–82 where more detailed comments on gene expression patterns are provided.
The next transition, the generation of iSchs from SCPs, also involves substantial changes in gene expression (Figure 40.2; Buchstaller et al., 2004; D’Antonio et al., 2006b; Dong et al., 1999; Frank et al., 1999). SCPs and iSchs also show a marked difference in survival regulation. SCPs are acutely dependent on axonal survival signals, chiefly NRG1 (Dong et al., 1995), whereas iSchs can prevent their own death by secretion of autocrine survival factors, a mechanism that SCPs lack (Dong et al., 1999; Meier et al., 1999). Because SCPs lack a basal lamina, formation of basal lamina by iSchs is another difference between these cells.
Cadherin 19 is initially activated when SCPs are formed from the crest but is downregulated in iSchs. It is therefore the only known gene that selectively marks the SCP stage (Takahashi and Osumi, 2005).
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780123972651000307
Tissue Engineering in Peripheral Nerve Regeneration
Xiaosong Gu, … Jie Liu, in Neural Regeneration, 2015
3.1.1 Sources of Schwann Cells
Schwann cells are the main glial cell in the PNS and play an essential role in the survival and functions of neurons. In response to nerve injury, Schwann cells undergo rapid changes in phenotype [199] and their basal lamina provides a conduit for axon regrowth, a critical process for nerve regeneration. Schwann cells also produce high levels of various growth factors [200–202] and participate in phagocytosis involved in the initial clearing of myelin debris. These characteristics make Schwann cells the first and most widely used support cells in peripheral nerve regeneration [203–205]. A genetic labeling technique has been used to trace Schwann cells after they are implanted into the nerve injury site, confirming their active roles in nerve regeneration [206,207].
In the 1980s, allogeneic Schwann cells were first selected as support cells for the repair of peripheral nerve injuries because allogeneic Schwann cells could support the peripheral nerve defect reconstruction without delay [208]. Because allogeneic Schwann cells are rejected by the host, immunosuppressive drugs must be given simultaneously. Without immunosuppression, foreign Schwann cells would be rejected, but the morphology of nerve grafts could be restored by host-derived Schwann cells. The most widely used immunosuppressive drugs are cyclosporin A and tacrolimus (FK506) [209–211].
Because of the immunological rejection involved in the use of allogeneic Schwann cells, the choice of syngeneic Schwann cells seems better. Researchers have compared the effects of allogeneic versus syngeneic Schwann cells in repairing a 10-mm rat sciatic nerve gap. Allogeneic Schwann cells were rejected by six weeks after implantation, when syngeneic Schwann cells were still identified. Allogeneic and syngeneic Schwann cells equally enhanced axonal regeneration but the quantity of axons was greater when using syngeneic Schwann cells [206].
Although immunosuppressive drugs can eliminate the rejection of allogeneic cells by the host, the clinical use of the drugs is still argued. Since the 1990s, autologous Schwann cells have been attempted as support cells. For example, nerve scaffolds seeded with autologous Schwann cells effectively reconstructed a rat median nerve gap and achieved outcomes equal to those by autologous nerve grafting [212]. With an addition of 106 autologous Schwann cells to the autologous venous graft, the length of the nerve gap that could be reconstructed increased from 30 to 60 mm in a rabbit model [213].
The issue of whether in vitro culture enables the expansion of Schwann cells to a certain number and the retention of Schwann cell functions is very important. Autologous Schwann cells can be obtained from the contralateral nerve or by enzymatic dissociation of predegenerated sciatic nerves followed by culture in a chemically defined medium. Predegeneration is considered a highly efficacious method to rapidly isolate Schwann cells without sacrifice of a healthy donor nerve or without a time-consuming process of cell culture and expansion [214–216]. Schwann cells divide in response to exogenous mitogens such as heregulin, forskolin, and cholera toxin. Although the mitogen-induced expansion leads to an abnormal phenotype of Schwann cells compared to normal Schwann cells, they can still form morphologically normal myelin sheaths around axons when introduced into a regenerating rat sciatic nerve [217–219].
Because isolation and culture of primary Schwann cells is a time-consuming process, immortalized Schwann cell lines—for example, SCTM41—have been taken into account. The SCTM41 cell line closely resembles primary Schwann cells. Axonal regeneration, however, is significantly reduced by SCTM41 cell-containing grafts compared to primary Schwann cell-containing grafts [220].
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128017326000057
Neuroglia, Overview
Angus M. Brown, Bruce R. Ransom, in Encyclopedia of the Human Brain, 2002
IV.B.1 Regulation of Nerve Development
Schwann cells have been shown to regulate nerve development. In Erbb3– mice the survival of neuronal populations, including DRG neurons, depends on trophic factors released from Schwann cell progenitors and immature Schwann cells. Candidates for this function are glial cell line derived neurotrophic factor (GDNF) and a related compound, neurturin. GDNF mRNA is found in Schwann cells and it is essential for motorneuron survival. Neurotroph 3 (NT3) is a possible candidate. It is formed by Schwann cells at birth and promotes survival and differentiation of early sensory neurons. Thus, NT3 seems to support DRG neurons. Additionally, DHH may help form fibroblasts, and Schwann cell survival and MAC may support axonal architecture.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B0122272102002430
Tissue engineering of the nervous system
Paul D. Dalton, … Alan R. Harvey, in Tissue Engineering (Third Edition), 2023
Schwann cells
Schwann cells can produce a variety of growth-promoting molecules that provide a growth substrate for injured CNS axons. In the injured spinal cord, Schwann cell transplants elicit axonal regeneration (see Section 17.9).38,54,57 Schwann cells also form myelin sheaths around CNS axons allowing signal conduction in regenerated CNS axons. In addition to axonal regeneration, an intraspinal Schwann cell graft promotes neuroprotection thereby reducing the additional loss of nervous tissue due to secondary events.38 Schwann cells grafted into the lesioned CNS show little or no tendency to migrate and integrate with the host tissues due to a mutual repulsion exhibited between Schwann cells and CNS-derived astrocytes, as demonstrated in vitro.33 Schwann cell transplants in the cystic cavity combined with maintaining an increased level of cyclic adenosine monophosphate result in improved functional recovery.58
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128244593000172
Neural Crest Diversification and Specification: Transcriptional Control of Schwann Cell Differentiation☆
M. Wegner, in Reference Module in Neuroscience and Biobehavioral Psychology, 2017
The Role of Transcription Factors in the Schwann Cell Lineage
Schwann cells and satellite glia are the two main glial cell types of the peripheral nervous system (PNS). Whereas satellite glia are found within ganglia in close association with neuronal somata, Schwann cells are found in close contact with axons in the peripheral nerves. Both Schwann cells and satellite glia arise from neural crest stem cells. Following their specification, Schwann cells go through several consecutive developmental stages, including the Schwann cell precursor and the immature Schwann cell, before they finally undergo terminal differentiation and become myelinating or nonmyelinating Schwann cells of the adult PNS (Jessen and Mirsky, 2005). Each developmental stage is characterized by specific patterns of gene expression, and progression from one stage into the next requires corresponding changes. Taking the role of transcription factors as regulators of gene expression into account, each stage of Schwann cell development and each transition from one stage into the next must be brought about by changes in transcription factor occurrence or activities.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128093245026201
Gastrointestinal Mesenchymal Tumors
David Papke MD, PhD, Leona Doyle MD, in Gastrointestinal and Liver Pathology (Third Edition), 2024
Prognosis and Therapy
Schwann cell hamartoma is benign and managed by polypectomy.
Mucosal Schwann Cell Hamartoma—Fact Sheet
Definition
- ■
-
Benign Schwann cell proliferation
Incidence and Location
- ■
-
Rare; arises in colonic mucosa
Gender, Race, and Age Distribution
- ■
-
Slight female predominance
Clinical Features
- ■
-
Incidental finding, small (<1 cm) mucosal polyp
Prognosis and Therapy
- ■
-
Benign tumor
Mucosal Schwann Cell Hamartoma—Pathologic Features
Microscopic Findings
- ■
-
Small proliferations of Schwann cells in the lamina propria that entrap colonic crypts
Immunohistochemistry
- ■
-
Diffuse expression of S-100 protein
- ■
-
Negative for C34, CD117, epithelial membrane antigen, smooth muscle actin, and claudin-1
Differential Diagnosis
- ■
-
Perineurioma
- ■
-
Neurofibroma
- ■
-
Ganglioneuroma
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780323527941000079
Peripheral Nervous System Topics
Enrico Marani, Egbert A.J.F. Lakke, in The Human Nervous System (Third Edition), 2012
Schwann Cells and Schwann Cell Lineage
Schwann cells are not only producing myelin in the PNS, but also play an important role in receptor functions and regeneration (for overview see Bunge, 1993). Schwann cell studies are still increasing due to proliferation and differentiation of Schwann cells and to their role in leprosy (Scollard, 2008). The possibility to culture Schwann cells (Martenson, 1992; Vleggeert-Lankamp, 2006) also enhanced Schwann cell studies.
Schwann cells originate as neural-crest cells (see section on “Embryology” above) and develop into Schwann cell precursor cells, associated with a large quantity of axons and characterized by GAP-43 (growth associated protein-43). The next stage is known by its expression of S100 and is subdivided by Zorick and Lemke (1996) into a committed and a premyelinated Schwann cell stage, in which multiple axon connection association until association with one axon respectively is the delineator. From these S100 stages promyelinating Schwann cells associated with one axon and a myelinated Schwann cell type also related to one axon with expression of Krox-20+ (= EGR-2; serum response zinc-finger protein) and MG, are discerned. Recently arguments in favor of the production of skin melanocytes by this myelinated Schwann cell type were brought forward (Adameyko et al., 2009). In the early process of enwrapping the axon, L1 and N-CAM are expressed on both axon and Schwann cell for homophilic contacts, afterwards these antigens disappear (Martini and Schachner, 1986).
Next to these two last types always prononmyelinating Schwann cells (characterized by SCIP+ = Oct-6/Tst-1 and related to multiple axons) and non-myelinating Schwann cells (related to multiple axons and characterized by CalC+ = galactocerebrocide) are present.
Promyelinated Schwann cells produce an extracellular matrix to begin the myelination process, which occurs after birth. These cells downregulate the embryologic/fetal Schwann cell genes and start transcription of the myelin genes. The non-myelinating Schwann cells stay to express immature gene characteristics after birth with some difference to embryologic/fetal expression (for detailed overviews see Zorick and Lemke, 1996; Jessen and Mirsky, 2002).
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780123742360100045
Молекулярная биология наследственных мотосенсорных полиневропатий
В конце 19 века Ж.-М. Шарко и П. Мари во Франции и Г.Г. Тус в Англии описали клинику наследственной мотосенсорной полиневропатии, которая была названа их именем: болезнь Шарко-Мари-Туса (Charcot-Marie-Tooth disease – CMT). Клинически более тяжелая полиневропатия с началом развития в раннем детстве была позднее описана двумя студентами Шарко – Ж. Дежерином и Ж. Соттом. Г. Русси и Г. Леви описали мотосенсорную полиневропатию с тремором движения. В течение последующих 50 лет последовало большое количество эпонимов, клинических описаний и громоздких, нефункциональных классификаций этих болезней.
Ситуация прояснилась только в 70-е и 80-е годы за счет развития электрофизиологии и генетики. Например, несколько групп исследователей измерили скорость проведения по нерву у пациентов с СМТ и обнаружили, что пациенты могут быть классифицированы на 2 группы независимо от типа наследования. Одни пациенты имели скорость проведения по нервам меньше чем 38 м/с, что предполагало первичную демиелинизацию как причину их полиневропатии (СМТ1). В другой группе скорость была более чем 38 м/с, и поэтому первичный аксональный процесс был более вероятной причиной их полиневропатии (СМТ2). Эти данные говорили о том, что имеются, по крайней мере, 2 различных патологических процесса развития СМТ.
За последние 10 лет появилось большое количество информации по новым генетическим формам СМТ. Эти данные открыли новые возможности исследования патофизиологии периферических полиневропатий и решения спорных вопросов их клиники. Например, синдром Русси-Леви, демиелинизирующая полиневропатия с тремором движения, обусловлен дупликацией 17р11.2 и точковыми мутациями гена миелинового гликопротеина Р(0) – myelin protein zero (MPZ), что характерно также и для наиболее распространенной демиелинизирующей формы СМТ – СМТ1А. Известно, что в одной семье с синдромом Русси-Леви могут быть пациенты с тремором движения и без. Это подтверждает, что синдром Русси-Леви является фенотипическим вариантом с вариабельной экспрессией СМТ1, а не отдельным заболеванием. Кроме того, синдром Дежерина-Сотта, тяжелая форма СМТ1 с ранним началом, обусловлен мутациями в одном из нескольких генов (ген периферического миелинового белка 22 – peripheral myelin protein 22 — PMP22, MPZ и ранний рост-отвечающий 2 ген – early growth response 2 — EGR2), этими же мутациями обусловлена и менее тяжелая полиневропатия СМТ1 с поздним началом. Тяжесть клиники полиневропатии зависит от природы специфической мутации, а не является результатом отдельного болезненного процесса. В новой классификации СМТ:
— СМТ1 – низкая скорость проведения по нервам и аутосомно-доминантный тип наследования;
— СМТХ – Х-сцепленный тип наследования;
— СМТ2 – аутосомно-доминантный тип наследования, нормальная скорость проведения по нерву и снижение амплитуды М-ответа и вызванного потенциала мышцы;
— СМТ4 – аутосомно-рецессивный тип наследования. Вышеуказанные группы разделены на подгруппы в зависимости от мутаций в генах белков шванновских клеток или нейронов.
Биология наследственных мотосенсорных полиневропатий.
Аксоны периферических нервов по всей длине покрыты шванновскими клетками. Во время развития предшественники шванновских клеток мигрируют с развивающимися периферическими аксонами. Незрелые шванновские клетки покрывают пучки развивающихся аксонов (этот процесс называется радиальная сортировка) и превращаются в миелинизирующие и немиелинизирующие клетки. Шванновские клетки, которые устанавливают связь с аксоном «один на один» (промиелинизирующая стадия развития шванновских клеток), начинают процесс миелинизации и становятся миелинизирующими шванновскими клетками. И, наоборот, шванновские клетки, которые не имеют такой ассоциации с аксоном, не активируют экспрессию гена миелина и становятся немиелинизирующими шванновскими клетками. Этот важный процесс контролируется аксонами, поэтому все незрелые шванновские клетки имеют возможность стать либо миелинизирующими, либо немиелинизирующими.
Первичная функция миелина – повысить скорость проведения импульса без значительного увеличения диаметра аксона. Эта функция достигается с помощью сальтаторного проведения, при котором нервные импульсы «прыгают» между электрически возбудимыми участками аксона (перехваты Ранвье), расположенными между электрически изолированными участками, покрытыми миелинизирующими шванновскими клетками. Так как большинство периферических нервов имеют пучки миелинизированных аксонов как большого, так и малого диаметра, их скорость проведения определяется, в основном, скоростью самых больших по диаметру миелинизированных волокон.
Недавние исследования миелинизированных аксонов и их перехватов Ранвье выявили удивительную сложность структуры. Миелиновая оболочка имеет 2 участка: компактный и некомпактный, каждый из которых содержит уникальный набор белков. Компактный участок содержит белки, которые формируют миелиновую оболочку в электрически изолированных участках аксона: MPZ, PMP22 и основной белок миелина. Некомпактный участок состоит из двух субдоменов: паранод и юкстапаранод (paranode and juxtaparanode). Паранодальный участок состоит из петель мембраны шванновских клеток и взаимодействующей с ними мембраны аксонов, прилегающей к перехвату Ранвье, и содержит белки шванновских клеток: MAG, коннексин 32, нейрофуцин 155 (connexin 32, neurofascin 155) и аксональные белки: Каспр, контактин (Caspr, contactin). Эти белки принимают участие во взаимодействии между шванновскими клетками и аксонами и в электрической изоляции нодального участка. Юкстапаранодальный участок имеет натриевые каналы и Каспр2 (Caspr2), экспрессирующиеся аксонами.
Знание процесса развития нерва, дифференциации шванновских клеток, миелинизации и формирования перехватов Ранвье облегчает понимание патогенеза всех типов полиневропатий, включая СМТ. Таким образом можно предполагать, что наследственные полиневропатии возникают вследствие мутаций, которые изменяют наиболее важные процессы развития нерва, например, нарушают связь между шванновскими клетками и их аксонами в паранодальном участке или процесс компакции миелина.
Мутации генов, кодирующих белки шванновских клеток.
СМТ1 и синдром Дежерина-Сотта вызваны мутацией в РМР22 и МРZ генах, коллирующих главные структурные белки компактного миелина. Компактный миелин содержит несколько структурных белков, включая 2 мембранных белка, MPZ(PO), основной белок миелина и РМР22. Последние два необходимы для нормальной миелинизации, их отсутствие у мышей приводит к значительным нарушениям компакции миелина и клинике невропатии. Мутации в МРZ и РМР22 генах у людей вызывают как СМТ1, так и более тяжелый по клинике синдром Дежерина-Сотта.
РМР22 (СМТ1А, синдром Дежерина-Сотта и наследственная невропатия со склонностью к параличам от сдавления).
Дупликация участка в 1,5мв на хромосоме 17, который включает ген, кодирующий РМР22, вызывает наиболее типичную форму СМТ – СМТ1А. Дупликация РМР22 гена в этой области является причиной болезни; мыши и крысы с экстратрансгенными копиями гена также имеют подобную демиелинизирующую невропатию. Начало клиники у пациентов с СМТ1А связано с появлением слабости и гипестезии в нижних конечностях в течение первых 10 лет жизни, позднее вовлекаются и верхние конечности. Хотя неизвестно, каким образом данная мутация вызывает невропатию, пациенты с СМТ1А имеют повышенное количество РМР22 в периферическом миелине, который в результате этого становится нестабильным.
Точечные мутации в РМР22, менее типичные, чем дупликации в гене, также могут вызывать СМТ1. Клинический фенотип пациентов с точечными мутациями РМР22 определяется природой мутации; у одних пациентов развивается невропатия, подобная той, которая вызывается дупликацией гена, у других же диагностируется более тяжелый синдром Дежерина-Сотта с началом в раннем неонатальном периоде. Всего две из известных точечных мутаций возникают в трансмембранных доменах РМР22. Молекулярные механизмы, с помощью которых РМР22 точечные мутации вызывают невропатию, неясны, но вероятно, они включают нарушение либо созревания белка либо белкового транспорта и таким образом, четко отличаются от вызывающих более типичную форму СМТ1А с дупликацией гена.
Делеция 1,5мв участка на хромосоме 17, дуплицированного при СМТ1, не вызывает СМТ, а приводит к асимметричной демиелинизирующей невропатии, называемой наследственной невропатией со склонностью к параличам от сдавления (ННСПС), характеризующейся преходящими эпизодами слабости в мышцах и потерей чувствительности. Снижение экспрессии РМР22 является причиной этого синдрома, так как он всегда выявляется у пациентов с мутацией в проксимальной части РМР22. Пациенты с ННСПС имеют в миелине участки плохой компактности с избыточными мембранными петлями, названными томакулами, которые ухудшают структурную целостность миелина, делая его более доступными для повреждения при внешней травме.
Таким образом, мутация в гене, кодирующем основной белок миелина, РМР22, может вызвать демиелинизирующую невропатию при помощи нескольких механизмов. Дупликация гена и повышенная экспрессия вызывают СМТ2А, тогда как делеция и пониженная экспрессия вызывают ННСПС. Точечные мутации могут вызывать ННСПС, СМТ1А или синдром Дежерина-Сотта в зависимости от природы мутации. Молекулярные механизмы при дупликации или делеции гена, вызывающие невропатию, вероятно лежат в основе нарушения структурной целостности компактного участка миелиновой оболочки. Молекулярные механизмы невропатии вследствие точечных мутаций РМР22 зависят от локализации специфической мутации и могут вовлекать изменения важных функций шванновских клеток, например, транспорта мембранного белка.
МРZ (СМТ1В и синдром Дежерина- Сотта).
Точечные мутации в гене, кодирующем главный структурный белок миелина, MPZ, могут вызывать как СМТ1В, клинически подобный СМТ1А, так и более тяжелый синдром Дежерина-Сотта. Мутации MPZ также были идентифицированы у пациентов с невыраженными симптомами невропатии и нечеткой демиелинизацией по данным электронейромиографии – “СМТ2 подобная” невропатия – и у нескольких пациентов с тяжелой невропатией с началом в пренатальном периоде, названной врожденной гипомиелинизирующей невропатией. Клинический спектр невропатий, вызываемых MPZ мутациями, таким образом, шире, чем вызываемых РМР22 мутациями. Спектр фенотипов, обусловленных мутациями в MPZ гене предполагает, что белок играет важную, но сложную роль в миелинизации. Одна из функций MPZ, подтвержденная множеством исследований, это медиация процесса компакции миелина посредством адгезии противопоставленных мембранных структур. MPZ – один из иммуноглобулинов супергенной семьи трансмембранных белков. Он действует как гомофильная молекула. Молекула адгезии во время экспрессии в гетерологичных клетках; экстраклеточная часть MPZ, экспрессированная на одной мембранной поверхности, может взаимодействовать с подобным участком этого же протеина, экспрессированного на другой мембранной поверхности. Также, анализ рентгеновской кристаллографии экстраклеточного домена показывает, что адгезия возникает за счет формирования взаимодействующих MPZ гомотетрамеров на противоположных мембранных поверхностях. Мыши, у которых экспрессия MPZ была инактивирована, имели некомпактные миелиновые оболочки и тяжелую периферическую невропатию. Поэтому, мутации в участках MPZ, ответственных за белок-белок взаимодействия, вызывают более тяжелый клинический фенотип, чем мутации в других участках. Разнообразие клинических фенотипов вследствие мутаций MPZ обусловлено, возможно, влиянием мутаций на MPZ – медиированный процесс гомофильной адгезии. Такие взаимодействия явно имеют важную роль в компакции миелина. MPZ также имеет регуляторную функцию в процессе миелинизации. Мыши, у которых экспрессия MPZ была инактивирована, имеют нарушенную регуляцию экспрессии гена миелина и нарушение расположения нескольких белков некомпактного миелина. Один из вероятных механизмов, при помощи которого MPZ регулирует процесс миелинизации, это через адгезию – медиированный каскад сигнальной трансдукции, который требует взаимодействия между цитоплазматическим доменом MPZ и другими компонентами клетки. Согласно этой гипотезе, мутации в цитоплазматическом домене MPZ сокращают MPZ – медиированную адгезию in vitro и вызывают наиболее тяжелые формы СМТ1В. Также, 14-аминокислотная последовательность в цитоплазматическом домене MPZ включает субстрат для протеинкиназы С, активность которой важна для процесса адгезии. Мутация аргинина в этом субстрате (R 198S) также вызывает СМТ1В.
Коннексин 32 (СМТХ).
Коннексин 32 (СХ32) – это белок, связывающий «дыры» в паранодальном участке и неровности шванновских клеток. Мутации в гене, кодирующем этот белок, выявляют у 10-15% пациентов с СМТ. Так как ген расположен на Х-хромосоме, то эта форма СМТ называется СМТХ. Клинический фенотип этих мутаций характеризуется слабостью и атрофией мышц и потерей чувствительности различной степени выраженности. Так как СХ32 убирает «дыры» между соседними петлями мембран шванновских клеток в паранодальной области, мутации в гене только слегка нарушают структуру компактного миелина. Согласно этой гипотезе, пациенты с СМТХ имеют почти нормальную скорость проведения по нерву и сильно сниженные амплитуды потенциала действия, что говорит больше о дегенерации аксона, чем о демиелинизации. Анализ образцов биопсии икроножного нерва пациентов с СМТХ подтверждает, что патология аксона является отличительной чертой при СМТХ. У мышей с мутацией СХ32 развивается невропатия, подобная невропатии пациентов с СМТХ, поэтому, очевидно, отсутствие функции СХ32 как связывающего «дыры» в паранодальном участке и шванновских клетках является серьезной причиной, чтобы вызвать аксональную дегенерацию. Неизвестно, как мутация вызывает аксональную дегенерацию, но механизм, очевидно, включает в себя нарушения клетка-клетка взаимодействий между паранодальными петлями мембраны шванновских клеток и связанными с ними аксонами. Разнообразие клинических фенотипов у пациентов с СМТХ, очевидно, обусловлено типом мутации и ее эффектом на функцию СХ32. Необходимо дальнейшее изучение функции СХ32 в шванновских клетках и патогенезе демиелинизации при СМТХ.
L – периаксин (СМТ4F).
Миссенс-мутации в гене, кодирующем L-периаксин, вызывают тяжелую аутосомную рецессивную демиелинизирующую невропатию СМТ4F. L-периаксин – специфичесикй белок шванновских клеток, связанный с мембраной. Подобно альтернативной изоформе S-периаксину L-периаксин имеет на своем карбоксильном конце PDZ домен, который необходим для взаимодействия с другими белками. Поэтому, очевидно, обе изоформы принимают участие в белок-белок взаимодействиях. L-периаксин экспрессируется на абаксональной поверхности миелинизирующих шванновских клеток, направленной не к аксону, а противопоставленной экстрацеллюлярному матриксу. Брофи и соавторы недавно сообщили о том, что L-периаксин формирует белковый комплекс с дистрофин-связанным белком 2 (DRP2) – цитоплазматическим белком, подобным мышечному белку дистрофину. Комплекс периаксин – DRP2 взаимодействует с цитоплазматическим доменом дистрогликана и связывается с ламинином 2 (мерозином), с компонентом базальной мембраны или с экстрацеллюлярным матриксом. L – периаксин необходим для функционирования DRP2 – дистрогликанового комплекса. Таким образом, L-периаксин формирует часть комплекса, подобного тому, который представлен в скелетной мышце и связывает базальную мембрану вне клетки и актиновый цитоскелет в клетке. Как в мышце, дистрофин-дистрогликановый комплекс, вероятно, стабилизирует внешнюю мембрану шванновских клеток. Известно, что взаимодействие шванновских клеток и базальной мембраны модулируют экспрессию гена миелина. Мутация гена, кодирующего белок L-периаксин, вызывает демиелинизирующую невропатию, так как нарушает взаимодействия шванновских клеток и базальной мембраны.
Хотя L-периаксин расположен в абаксональной мембране миелинизирующих шванновских клеток, его субклеточная локализация менялась во время развития. В эмбриональных шванновских клетках L-периаксин находится в ядре, тогда как в перинатальном периоде он находится в адаксональной или периаксональной цитоплазме шванновских клеток. Гистология периферического нерва пациентов с СМТ4F показывает структурные нарушения в паранодальной области с «разрывами» между паранодальными петлями и прилегающим аксоном. Предполагают, что эти изменения вызываются нарушением функции периаксина в периаксональной области.
Ранний рост-отвечающий 2 белок – EGR2 (CMT1).
Несколько групп исследователей недавно определили мутации в гене, кодирующем фактор транскрипции EGR2 (или KROX20), как причину новой формы доминантно наследуемой демиелинизирующей невропатии.
Экспрессия EGR2 начинает регулироваться в шванновских клетках на стадии промиелинизации, когда устанавливаются один-на-один отношения с аксонами, и наиболее высока экспрессия в миелинизирующих шванновских клетках. Также EGR2 и его матричная РНК являются продуктами и других главных генов миелина. Как на примере и других генов миелина, экспрессия EGR2 требует взаимодействия между шванновскими клетками и аксонами, поэтому он исчезает после трансекции нерва и восстанавливается во время реиннервации. Мышцы с недостаточной экспрессией EGR2 имеют демиелинизирующую периферическую невропатию, при которой шванновские клетки перестают развиваться на промиелинизирующей стадии развития, поэтому, очевидно, экспрессия EGR2 необходима для минования этой стадии. Последние исследования также показали, что EGR2 может регулировать несколько генов миелина в шванновских клетках, и что мутации в EGR2 вызывают невропатию вследствие нарушения экспрессии этих генов.
Инактивации гена, кодирующего фактор транскрипции РОИ-домен (Oct 6) у мышей, также приводит к остановке развития шванновских клеток на промиелинизирующей стадии, подобно тому, что наблюдается у животных с недостаточной экспрессией EGR2. Однако, остановка развития шванновских клеток при недостаточной экспрессии Oct 6 является временной, и периферические нервы миелинизируются нормально. Экспрессия Oct 6 в шванновских клетках начинается до экспрессии EGR2, достигает наивысшего развития в промиелинизирующую стадию и снижается в миелинизирующих шванновских клетках. Так как EGR2 экспрессируется в шванновских клетках мышей с недостаточной экспрессией Oct 6 во время стадии миелинизации, значит экспрессия EGR2 может проходить без экспрессии Oct 6. Эти данные предполагают, что Oct 6 руководит экспрессией EGR2 во время развития шванновских клеток и что Oct 6 может играть роль в своевременном начале экспрессии EGR2 и миелинизации. Мутации в Oct 6, вызывающие СМТ, не были определены, вероятно, из-за преходящей природы дефекта периферической миелинизации.
Миотубулярин- связывающая фосфатаза 2 (СМТ4В).
Мутация в гене, кодирующем миотубулярин – связывающую фосфатазу 2 (MTMR2), приводит к СМТ4В, рецессивно наследуемой демиелинизирующей невропатии, которая также называется наследственная моторная и сенсорная невропатия с фокально сложенными миелиновыми оболочками. MTMR2 – один из семьи белков, характеризующийся последовательностью из 10 аминокислот, схожий с активным сайтом как тирозин-, так и серинфосфатазы, которые, таким образом, являются двойственно-специфическими фосфатазами. Мутация в миотубулярине вызывает миотубуляриновую миопатию. Недавние исследования миотубулярина предполагают, что он отнимает фосфатазу от фосфатидил-инозитол-3-монофосфатазы. И, таким образом, вовлекается в регуляцию каскада сигнальной трансдукции фосфатидилинозитол-3-киназы. Хотя функция MTMR2 в периферической нервной системе неясна, образцы биопсии икроножного нерва показывают сегментарную демиелинизацию, связанную с избыточными петлями миелина, что предполагает, что MTMR2 может играть роль в регуляции «обертывания» миелина.
Мутации в генах, кодирующих нейрональные белки.
Как уже видно, разные нарушения процесса миелинизации шванновских клеток могут вызвать СМТ. Периферическая невропатия также может быть вызвана, однако, нарушениями функции аксона, вероятно, ввиду прекращения аксонального транспорта. Мутации в трех генах, кодирующих экспрессию белков в нейронах, обсуждаются ниже.
Легкая цепь нейрофиламента (СМТ и СМТ2Е).
Миссенс-мутации в легкой цепи нейрофиламента (NEFL), представленные в нейронах, вызывают наследственную невропатию СМТ2Е. Пациенты с СМТ2 не имеют снижения скорости проведения по нерву, что предполагает, что первичной причиной невропатии является патология аксона. Первые пациенты с мутациями NEFL и невропатией имели клинику, типичную для СМТ2: мышечная слабость и снижение чувствительности, нормальная скорость проведения по нерву, снижение амплитуды потенциала действия двигательных и чувствительных нервов. ДеДжонг и соавторы недавно описали бельгийскую семью с мутацией NEFL, снижением скорости проведения по нерву и фенотипом СМТ1. Согласно этой находке, мыши с недостаточной экспрессией NEFL также имеют существенное снижение скорости проведения по нерву (10 м/с) с резкой атрофией нервов, хотя клинический фенотип выражен умеренно. Эти данные предполагают, что мутации в гене NEFL, экспрессированном в нейронах, могут вызвать как СМТ2 с нормальной скоростью проведения по нерву, так и СМТ1 со сниженной скоростью. Классификация невропатии как демиелинизирующей, либо аксональной в зависимости от скорости проведения по нерву не всегда может отражать молекулярную патофизиологию процесса болезни.
KIF1Bβ (СМТ2А).
Недавно была выявлена семья с СМТ2 миссенс-мутацией в β-изоформе гена, кодирующего кинезин KIF1B-белок, экспрессированный в нейронах и участвующий в микротубулярном аксональном транспорте. Невропатия в этой семье наследуется аутосомно-доминантно и, на основании связи с хромосомой 1, классифицируется как СМТ2А. Мутация кинезина в этой семье изменяет амино-терминальную часть белка, в связи с чем нарушается связь с микротубулами и транспорт органелл по аксону. Согласно этим данным, мыши с одним из двух кинезин KIF1B генов, инактивированным гомологичной рекомбинацией, также имеют аксональную периферическую невропатию. Транспорт синаптотагмина, компонента для будущих синаптических везикул, снижен в дистальной части аксона у этих животных, поэтому неспособность транспортировать компоненты синаптических везикул может быть причиной аксональной невропатии. Все вышеупомянутые данные говорят о том, что нарушение аксонального транспорта может вызвать периферическую невропатию.
Гигаксонин (GAN).
Гигантская аксональная невропатия (GAN) – редкое аутосомно-рецессивное заболевание, обусловленное, по последним данным, миссенс-мутациями в новом белке цитоскелета гигаксонине, экспрессированном в нейронах. В периферических нервах пациентов с этим заболеванием находят, чаще в области перехватов Ранвье, увеличение калибра аксона за счет масс из тесно скомпанованных нейрофиламентов. Эти изменения свидетельствуют о тяжелой аксональной периферической невропатии. Нарушения в организации нейрофиламентов находят и в головном мозге этих пациентов, что, вероятно, является причиной задержки умственного развития. Пациенты с гигантской аксональной невропатией имеют нарушение структуры других филаментов, например, филаментов волос, что приводит к характерному симптому спутанных волос. Хотя мутация в гене гигаксонина вызывает явную аксональную патологию, молекулярная природа этих изменений пока еще неясна. Подобно мутациям в генах легкой цепи нейрофиламента и кинезина KIF1β, мутация в гене гигаксонина, вероятно, нарушает аксональный транспорт.
Мутации в гене, кодирующем ганглиозид-индуцированный, связанный с дифференциацией, белок 1 (СМТ4А).
Мутации в гене, кодирующем новый белок с неизвестной функцией – ганглиозид-индуцированный, связанный с дифференциацией, белок 1 (GDAP1), вызывают рецессивно-наследуемую СМТ4А. Мутации могут вызывать либо демиелинизирующие, либо аксональные невропатии. Исследования матричных РНК предполагают, что ген GDAP1 экспрессируется в шванновских клетках, хотя первоначально он был определен в линии нейронов. Таким образом, еще неизвестно, чьи функции нарушают мутации GDAP1 — шванновских клеток или нейронов, или и тех, и других. Одно вероятно, что мутации GDAP1 прекращают взаимодействия между шванновскими клетками и аксонами.
Роль аксональной дегенерации.
Хотя демиелинизация является патофизиологической чертой СМТ1, клинические симптомы этого заболевания – мышечная слабость и потеря чувствительности – обусловлены аксональной дегенерацией, а не демиелинизацией. Например, у детей с СМТ1 диагностируют сниженную скорость проведения по нерву до развития симптомов, а затем, с прогрессированием заболевания, скорость изменяется незначительно. Таким образом, вероятно, не демиелинизация вызывает клинику заболевания. В дополнение к этому, Кражевский с соавторами доказали, что данные амплитуд потенциала действия двигательных нервов, а не скорости проведения по нерву коррелируют с выраженностью мышечной слабости у пациентов с СМТ1А. Это предполагает, что аксональная дегенерация является причиной мышечной слабости. Наконец, существует и анатомическое доказательство прогрессирующей потери аксонов при СМТ1, а также и у мышей, избыточно экспрессирующих РМР22 (животная модель СМТ1А). Все эти данные дают веские основания полагать, что дистальная аксональная дегенерация, а не демиелинизация является основной причиной клиники при СМТ1.
Несколько исследований показали, что при демиелинизирующих периферических невропатиях, включая СМТ1, нарушаются взаимодействия между шванновскими клетками и аксонами, что вызывает значительные изменения в физиологии аксона. Например, мыши с демиелинизирующей периферической невропатией (как при СМТ1А), вызванной точечной мутацией РМР22, имеют значительные нарушения и структуры аксона, и его функции, включая изменения в нейрофиламентной фосфорегуляции, повышение плотности нейрофиламентов и снижение аксонального транспорта. Подобные изменения были найдены у пациентов с СМТ1. Нарушения взаимодействия шванновских клеток с аксонами, которые возникают при СМТ1 и других демиелинизирующих периферических невропатиях, приводят к изменениям в плотности «упаковки» нейрофиламентов, в фосфориляции, аксональном транспорте, отсюда возникает аксональная дегенерация. Вызывают ли эти изменения повреждения аксона напрямую или являются маркерами какой-то другой, более фундаментальной патологии аксона, пока не известно.
Хотя также еще неясно, какова природа “сигнала” от шванновских клеток, который модулирует плотность “упаковки” нейрофиламентов, фосфориляцию нейрофиламентов и аксональный транспорт, но она, вероятно, изменена или модифицирована в дисмиелинизирующих шванновских клетках. Этот “сигнал”, возможно, является частью скоординированной программы экспрессии гена миелина, так как нормальными шванновскими клетками он тоже производится. Самое удачное место для начала проводящего «сигнал» пути – это паранодальный участок миелинизирующих шванновских клеток, где и в шванновской клетке, и в ее аксолемме имеются для этого некоторые особенности. Дальнейшее изучение молекулярной архитектуры паранода и окружающей его аксолеммы должно способствовать пониманию как молекулярного строения проводящих путей, с помощью которых сообщаются шванновские клетки и аксоны, так и пониманию аксональной дегенерации при демиелинизирующей невропатии.
Заключение.
Тот факт, что дисмиелинизирующие шванновские клетки могут вызывать вторичное повреждение аксона, является важным для лечения СМТ, так как клиника СМТ в основном обусловлена этим повреждением аксона. Регуляция миелинизации сложна, и многочисленные мутации по-разному влияют на физиологию шванновских клеток. По этой причине коррекция дефекта шванновской клетки при СМТ будет также сложной, и могут потребоваться различные мероприятия для разных типов мутаций. Однако, аксональную дегенерацию при СМТ можно предотвратить с помощью достаточной дозы экзогенного фактора роста, например, цилиарного нейротрофического фактора или глия-производного нейротрофического фактора роста, которые, как известно, эффективны при дегенерации моторных нейронов. Этот подход к лечению может быть эффективен для всех типов СМТ, не учитывая вид мутации и характер молекулярной патофизиологии болезненного процесса. Также он может предотвратить развитие неврологической клиники, если препарат дается на ранних этапах болезни или уменьшить выраженность клиники за счет аксональной дегенерации, если препарат дается на поздних этапах. Дальнейшее изучение того, каким образом миелинизирующие шванновские клетки влияют на их аксоны, и как развивается аксональная регенерация, или как предотвратить аксональную дегенерацию, является, таким образом, наиболее важным для дальнейшего развития рациональной молекулярной терапии СМТ.
текст: С.В.Копишинская, А.В.Густов
Кафедра неврологии, психиатрии и наркологии ФПКВ Нижегородской государственной медицинской академии
Очень часто при описании нервной системы используются «электрические» термины: например, нервы сравниваются с проводами. Это потому, что по нервному волокну действительно перемещается электрический сигнал. Каждому из нас известно, что оголенный провод опасен, ведь он бьет током, и по этой причине люди пользуются изоляционными материалами, не проводящими электричество. Природе тоже не чужда техника безопасности, и нервные «провода» она обматывает своим собственным изолирующим материалом — миелином.
Сложная обёртка
Миелин окружает отростки нервных клеток, изолируя их от внешнего воздействия. Это необходимо для более надежной и быстрой передачи сигнала по нервной системе. Благодаря изоляции нервного волокна электрический сигнал не рассеивается и добирается до места назначения без помех. Скорость прохождения сигнала по миелиновым и безмиелиновым волокнам может отличаться на три порядка: от 70 до 140 м/с и от 0,3 до 0,5 м/с соответственно.
По сути миелин — это клеточная мембрана глиальных клеток, многократно обмотанная вокруг аксона. Сама мембрана на 70–75% состоит из липидов и на 25–30% — из белков. В периферической нервной системе донором мембран становятся шванновские клетки, а в центральной — олигодендроциты. Эти клетки бережно обматывают своими мембранами ценные каналы связи, чтобы обеспечить надежное взаимодействие нервной системы и периферических органов. Миелин покрывает нервное волокно не целиком: существуют промежутки между наслоениями миелина, называемые перехватами Ранвье (рис. 1). Есть прямая зависимость между расстоянием от одного промежутка до другого и скоростью распространения нервного импульса по волокну: чем больше расстояние между перехватами Ранвье, тем выше скорость передачи сигнала в нерве [1].

Рисунок 1. Нервное волокно, обернутое миелином. Видны ядра шванновских клеток (nucleus of Schwann cell) и перехваты Ранвье (nodes of Ranvier) — участки аксона, которые не покрыты миелиновой оболочкой.
Если говорить о белках, входящих в состав миелина, то надо уточнить, что это не только простые белки. В миелине встречаются гликопротеины — белки, к которым присоединены короткие углеводные последовательности. Важной составляющей миелина является главный структурный белок миелина (myelin basic protein, MBP), впервые выделенный около 50 лет назад. MBP — это трансмембранный белок, который может многократно «прошивать» липидный слой клетки. Его различные изоформы (рис. 2) кодируются геном под названием Golli (gene in the oligodendrocyte lineage). Структурной основой миелина служит изоформа массой 18,5 килодальтон [2].

Рисунок 2. Различные изоформы основного белка миелина (MBP) создаются на основе одного и того же гена. Например, для синтеза изоформы массой 18,5 кДа используются все экзоны, кроме экзона II.
В состав миелина входят сложные липиды цереброзиды. Они представляют собой аминоспирт сфингозин, соединенный с жирной кислотой и остатком углевода. В синтезе липидов миелина принимают участие пероксисомы олигодендроцитов. Пероксисомы — это липидные пузырьки с различными ферментами (в общей сложности известно около 50 видов пероксисомных энзимов). Эти органеллы занимаются, в частности, β-окислением жирных кислот: жирных кислот с очень длинной цепью (very long chain fatty acids, VLCFA), некоторых эйкозаноидов и полиненасыщенных жирных кислот (ПНЖК, polyunsaturated fatty acids, PUFAs). Поскольку миелин может содержать до 70% липидов, пероксисомы крайне важны для нормального метаболизма этого вещества. Они используют N-ацетиласпартат, вырабатываемый нервной клеткой, для постоянного синтеза новых липидов миелина и поддержания его существования. Кроме этого, пероксисомы принимают участие в поддержании энергетического метаболизма аксонов [3].
Важная обёртка
Миелинизация (постепенная изоляция нервных волокон миелином) начинается у людей уже в эмбриональном периоде развития. Первыми этот путь проходят подкорковые структуры. В течение первого года жизни происходит миелинизация отделов периферической и центральной нервной системы, отвечающих за двигательную активность. Миелинизация участков головного мозга, регулирующих высшую нервную деятельность, заканчивается к 12–13 годам. Из этого видно, что миелинизация тесно связана со способностью отделов нервной системы осуществлять специфические для них функции. Вероятно, именно активная работа волокон до рождения запускает их миелинизацию.
Дифференцировка клеток — предшественниц олигодендроцитов зависит от ряда факторов, связанных с работой нейронов. В частности, работающие отростки нейронов могут выделять белок нейролигин 3, который способствует пролиферации и дифференциации клеток-предшественниц [4]. В дальнейшем созревание олигодендроцитов происходит за счет ряда других факторов. В статье с характерным названием «Насколько велик миелинизирующий оркестр?» описывается происхождение олигодендроцитов в разных частях мозга [5]. Во-первых, в различных частях мозга олигодендроциты начинают созревать в разное время. Во-вторых, за их созревание отвечают разные клеточные факторы, что тоже зависит от региона нервной системы (рис. 3). У нас может возникнуть вопрос: а сходны ли между собой олигодендроциты, появившиеся с таким расхождением в стартовых данных? И насколько схож у них миелин? В целом, авторы статьи считают, что между популяциями олигодендроцитов из разных участков головного мозга действительно существуют различия, и обусловлены они во многом именно местом закладки клеток, воздействием на них окружающих нейронов. И всё же типы миелина, синтезируемые разными пулами олигодендроцитов, не имеют настолько больших отличий, чтобы они не были взаимозаменяемыми.

Рисунок 3. Различия во времени закладки олигодендроцитов в разных отделах головного мозга и в клеточных факторах, влияющих на их развитие.
Сам процесс миелинизации нервных волокон в центральной нервной системе происходит следующим образом (рис. 4). Олигодендроциты выпускают несколько отростков к аксонам разных нейронов. Входя с ними в контакт, отростки олигодендроцитов начинают оборачиваться вокруг них и расползаться по длине аксона. Количество оборотов постепенно увеличивается: в некоторых участках ЦНС их число доходит до 50. Мембраны олигодендроцитов становятся всё более тонкими, распространяясь по поверхности аксона и «выдавливая» из себя цитоплазму. Чем раньше слой миелина был обернут вокруг нервного окончания, тем более тонким он будет. Самый внутренний слой мембраны остается довольно толстым — для осуществления метаболической функции. Новые слои миелина наматываются поверх старых, перекрывая их так, как показано на рисунке 4 — не только сверху, но и увеличивая площадь аксона, покрытую миелином.

Рисунок 4. Миелинизация нервного волокна. Мембрана олигодендроцита наматывается на аксон, постепенно уплотняясь с каждым оборотом. Внутренний, прилегающий к аксону слой мембраны остается относительно толстым, что необходимо для выполнения метаболической функции. На разных частях рисунка (а-в) с разных ракурсов показано постепенное наматывание новых слоев миелина на аксон. Красным цветом выделен более толстый, метаболически активный слой, синим — новые уплотняющиеся слои. Внутренний слой миелина (inner tongue на части б) охватывается всё новыми и новыми слоями мембраны не только сверху, но и по бокам (в), вдоль аксона.
Миелинизация нервных волокон олигодендроцитами также значимо зависит от белка нейрегулина 1. Если он не воздействует на олигодендроциты, то в них запускается программа миелинизации, не учитывающая активность нервной клетки. Если же олигодендроциты получили сигнал от нейрегулина 1, то далее они начнут ориентироваться на работу аксона, и миелинизация будет зависеть от интенсивности выработки глутамата и активации им специфических NMDA-рецепторов на поверхности олигодендроцитов [6]. Нейрегулин 1 — ключевой фактор для запуска процессов миелинизации и в случае шванновских клеток [7].
Изменчивая обёртка
Миелин постоянно образуется и разрушается в человеческом организме. На синтез и распад миелина могут влиять факторы, связанные с особенностями внешней среды. Например, воспитание. С 1965 по 1989 год Румынией руководил Николае Чаушеску. Он установил жесткий контроль над репродуктивным здоровьем и институтом брака в своей стране: усложнил процедуру развода, запретил аборты и ввел ряд стимулов и льгот для женщин, имевших более пяти детей. Итогом этих мер стало ожидаемое повышение рождаемости. Вместе с рождаемостью увеличилось количество криминальных абортов, не добавивших здоровья румынкам, и возросло количество детей-отказников. Последние воспитывались в детских домах, где с ними не очень-то активно общался персонал. Румынские дети в полной мере ощутили на себе то, что называется социальной депривацией — лишение возможности полноценного общения с другими людьми. Если речь идет о маленьком ребенке, то следствиями социальной депривации станут нарушение формирования эмоциональных привязанностей и расстройство внимания. Когда режим Чаушеску пал, западным ученым предстояло в полной мере оценить результат социальной политики этого диктатора. Румынских детей, имеющих выраженные проблемы со вниманием и установкой социальных контактов, впоследствии стали называть детьми Чаушеску.
Кроме различий при выполнении нейропсихологических тестов, у детей Чаушеску по сравнению с детьми, не находившимися в таких условиях, отличалось даже строение головного мозга [8]. При оценке состояния белого вещества мозга ученые используют показатель фрактальной анизотропии. Он позволяет оценить плотность нервных волокон, диаметр аксонов и их миелинизацию. Чем больше фрактальная анизотропия, тем разнообразнее волокна, которые встречаются в этой области мозга. У детей Чаушеску отмечалось снижение фрактальной анизотропии в пучке белого вещества, соединяющего височную и лобную доли в левом полушарии, то есть связи в этом регионе были недостаточно сложными и разнообразными, с нарушениями миелинизации. Такое состояние связей мешает нормальному проведению сигналов между височной и лобной долями. В височной доле находятся центры эмоционального реагирования (миндалина, гиппокамп), а орбитофронтальная кора лобной доли также связана с эмоциями и принятием решений. Нарушение формирования связей между этими отделами мозга и проблемы в их работе в итоге приводили к тому, что выросшие в детдомах дети испытывали трудности в установлении нормальных отношений с другими людьми.
На миелинизацию также может влиять и состав еды, которую дают ребенку. При белково-энергетической недостаточности питания отмечается снижение образования миелина. Недостаток жирных кислот тоже отрицательно сказывается на синтезе этого ценного вещества, так как оно больше чем на 2/3 состоит из липидов. Дефицит железа, йода и витаминов группы В приводит к снижению образования миелина [9]. В основном эти данные были получены при изучении лабораторных животных, но история, к сожалению, дала людям возможность оценить влияние недостатка еды и на формирующийся мозг ребенка [10]. Голодная зима (голл. hongerwinter) 1944–1945 гг. в Нидерландах привела к тому, что родилось множество детей, чьи матери плохо питались. Оказалось, что в условиях голодания мозг этих детей формировался с нарушениями. В частности, наблюдалось большое количество нарушений именно в белом веществе, то есть возникали проблемы с формированием миелина. В итоге это приводило к разнообразным психическим расстройствам.
Поврежденная обёртка

Рисунок 5. Нарушение чувствительности по полиневритическому типу. Название «носки — перчатки» связано с тем, что анатомические зоны, соответствующие поражению нервов, похожи на области, покрываемые этими предметами одежды.
Как мне кажется, для человеческого организма вполне подходит следующее правило: если есть орган, значит, к нему должна быть болезнь. В принципе, это правило можно расширить до молекулярных процессов: есть процесс — есть и болезни, связанные с нарушением этого процесса. В случае с миелином это демиелинизирующие заболевания. Их довольно много, но подробнее я расскажу о двух — синдроме Гийена-Барре и рассеянном склерозе. При этих расстройствах повреждение миелина приводит к нарушению адекватного проведения сигнала по нервам, что и обуславливает симптомы болезни.
Синдром Гийена-Барре (СГБ) — это заболевание периферической нервной системы, при котором происходит разрушение миелиновой оболочки, формируемой шванновскими клетками. СГБ является классическим аутоиммунным заболеванием. Как правило, ему предшествует инфекция (часто — вызванная микробом Campylobacter jejuni). Присутствие различных возбудителей в организме человека запускает аутоиммунное повреждение миелина нервных волокон T- и B-лимфоцитами. Клинически это проявляется мышечной слабостью, нарушением чувствительности по типу «носки — перчатки» (полиневритический тип) (рис. 5). В дальнейшем мышечная слабость может нарастать вплоть до полного паралича конечностей и поражения туловищной мускулатуры. Поражения чувствительной нервной системы также могут быть разнообразны: от снижения способности различать собственные движения (нарушение глубокой чувствительности) до выраженного болевого синдрома. При тяжелых формах СГБ главную опасность представляет потеря способности к самостоятельному дыханию, требующая подключения к аппарату искусственной вентиляции легких (ИВЛ). Для лечения СГБ в настоящее время используют плазмаферез (очистку плазмы от вредных антител) и внутривенные вливания препаратов человеческого иммуноглобулина для нормализации иммунного ответа. В большинстве случаев лечение приводит к стойкому выздоровлению.
Рассеянный склероз (РС) заметно отличается от СГБ. Во-первых, это демиелинизирующее заболевание приводит к поражению центральной нервной системы, то есть затрагивает миелин, синтезируемый олигодендроцитами. Во-вторых, с причинами РС до сих пор много неясного: слишком большое разнообразие генетических и средовых факторов задействовано в патогенезе заболевания. Принципиальный момент в запуске РС — нарушение непроницаемости гематоэнцефалического барьера (ГЭБ) для иммунных клеток. В норме ткань мозга отгорожена от всего остального организма этим надежным фильтром, который не пропускает к ней множество веществ и клеток, в том числе иммунных. ГЭБ появляется уже в эмбриональном периоде развития, изолируя ткань мозга от формирующейся иммунной системы. В это время иммунная система человека «знакомится» со всеми существующими тканями, чтобы в дальнейшем, при взрослой жизни, не нападать на них. Мозг и ряд других органов остаются «не представленными» иммунной системе. При нарушении целостности ГЭБ иммунные клетки получают возможность для атаки незнакомых ей тканей мозга. В-третьих, РС отличается более тяжелыми симптомами, которые требуют других терапевтических подходов. Симптоматика зависит от того, где локализуются повреждения нервной системы (рис. 6 и 7). Это может быть шаткость походки, нарушения чувствительности, различные когнитивные симптомы. Для лечения РС используются высокие дозы глюкокортикоидов и цитостатики, а также препараты интерферона и специфические антитела (натализумаб). По-видимому, в дальнейшем будут развиваться новые методы лечения РС, основанные непосредственно на восстановлении миелиновой оболочки в поврежденных участках мозга. Ученые указывают на возможность трансплантации клеток — предшественниц олигодендроцитов или усиления их роста за счет введения инсулиноподобного фактора роста или тиреоидных гормонов [11]. Однако это еще впереди, а пока неврологам недоступны более «молекулярные» методы лечения.

Рисунок 6. Очаги поражения центральной нервной системы при рассеянном склерозе на МРТ выглядят как белые бляшки.

Рисунок 7. В зависимости от места поражения нервной системы при рассеянном склерозе может быть разная симптоматика: от тремора и атаксии при повреждении мозжечка до эмоциональных расстройств при локализации очагов в лобных долях.
- Wu L.M., Williams A., Delaney A., Sherman D.L., Brophy P.J. (2012). Increasing internodal distance in myelinated nerves accelerates nerve conduction to a flat maximum. Curr. Biol. 22, 1957–1961;
- Harauz G. and Boggs J.M. (2013). Myelin management by the 18.5-kDa and 21.5-kDa classic myelin basic protein isoforms. J. Neurochem. 125, 334–361;
- Kassmann C.M. (2014). Myelin peroxisomes — essential organelles for the maintenance of white matter in the nervous system. Biochemie. 98, 111–118;
- Venkatesh H.S., Johung T.B., Caretti V., Noll A., Tang Y., Nagaraja S. et al. (2015). Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell. 161, 803–816;
- Tomassy G.S. and Fossati V. (2014). How big is the myelinating orchestra? Cellular diversity within the oligodendrocyte lineage: facts and hypotheses. Front. Cell Neurosci. 8, 201;
- Michalski J.-P. and Kothary R. (2015). Oligodendrocytes in a nutshell. Front. Cell Neurosci. 9, 340;
- Salzer J.L. (2012). Axonal regulation of Schwann cell ensheathment and myelination. J. Peripher. Nerv. Syst. 17, 14–19;
- Eluvathingal T.J., Chugani H.T., Behen M.E., Juhász C., Muzik O., Maqbool M. et al. (2006). Abnormal brain connectivity in children after early severe socioemotional deprivation: a diffusion tensor imaging study. Pediatrics. 117, 2093–2100;
- Prado E.L. and Dewey K.G. (2014). Nutrition and brain development in early life. Nutr. Rev. 72, 267–284;
- Hulshoff Pol H.E., Hoek H.W., Susser E., Brown A.S., Dingemans A., Schnack H.G. et al. (2000). Prenatal exposure to famine and brain morphology in schizophrenia. Am. J. Psychiatry. 157, 1170–1172;
- Bhatt A., Fan L.W., Pang Y. (2014). Strategies for myelin regeneration: lessons learned from development. Neural. Regen. Res. 9, 1347–1350.